Michael R. Hunsaker, PhD
Center for Integrative Neuroscience Human Behavior; University of Utah; Salt Lake City, UT, USA
Abstract
With the increasing sophistication of the genetic techniques used to develop mouse models of genetic disorders, it is imperative that the techniques used to elucidate the behavioral phenotypes of these models evolve just as rapidly. At present, mouse models developed to study neurodevelopmental disorders either demonstrate inconsistent phenotypes or lack behavioral phenotypes when tested using the standard battery of behavioral tasks. In this chapter I describe a behavioral endophenotyping approach that allows researchers to explicitly model in mice the clinical phenotypes reported in human genetic disease. This approach facilitates a comprehensive approach to studying the effects of genetic mutations on behavior by individually evaluating each of the different domains / attributes of memory (e.g., time, space, sensory / perceptual, response, affect, and language), as well as social behaviors and executive function. The data obtained from this approach can be translated back to the clinical population on a per attribute basis, allowing for a dialogue between the clinic and basic science to facilitate the generation of testable hypotheses. A reciprocal interaction across levels of analysis also results in efficient development of outcome measures that can be used to evaluate the efficacy of treatment or interventional studies in the mouse model that potentially show predictive validity with later clinical trials. Examples of standardizing the behavioral phenotyping process for human disease using the NIH Toolbox, a collection of cognitive assessments, as well as a proposed murine analog of the NIH Toolbox are provided to illustrate the power of applying the attribute model to behavioral phenotyping.
Introduction
With the increasing sophistication of the genetic techniques used to develop mouse models of genetic disorders, it is imperative that the techniques used to elucidate the behavioral phenotype of these models evolve just as rapidly. Although there is a movement toward adopting standardized behavioral phenotyping protocols, to a large degree neuroscientists evaluating mouse models of genetic disorders still lack sensitive behavioral assays needed to evaluate the core cognitive deficits present in genetic disorders. At present, mouse models, particularly those developed to study neurodevelopmental or other genetic disorders, demonstrate inconsistent phenotypes or entirely lack behavioral phenotypes when tested using the most common behavioral tasks: including the water maze, Barnes maze, active / passive avoidance, rotarod, or fear conditioning (Baker et al., 2010; Bohlen et al., 2009; Cannon & Keller, 2006; Hasler et al., 2006; Kendler & Neale, 2010; Long et al., 2006; Manji et al., 2003; Paylor & Lindsay 2006; Rustay et al., 2003; Spencer et al., 2011; Weiser et al., 2005; Yan et al., 2004). Furthermore, it has been shown that even extremely subtle differences between individual laboratory protocols for these common tasks changes observed phenotypes (e.g., differences in care taken while fixing a rough surface to a rotarod has been shown to dissociate performance across collaborating laboratories studying the same mouse strain; cf., Crabbe et al., 1999; Crabbe & Wahlsten, 2003; Wahlsten, 1972a, 2001; Wahlsten et al., 2003a,b,c,d,e, 2006).
Additionally, mouse models often demonstrate phenotypes that are not specifically associated with any genetic disorder in particular, but are more aptly described as shared clinical phenotypes that similarly present across a wide array of disorders (e.g., gross learning and memory deficits, anxiety, depression). The interpretation of such inconclusive findings is often that the mouse model fails to recapitulate the phenotypes observed in patients (cf., Gottesman & Gould, 2003; Gould & Gottesman, 2006; Weiser et al., 2005). Unfortunately, these types of findings are analogous to inconsistent findings in clinical populations when standardized neuropsychological tests are administered -- many different populations show very similar deficits despite nonoverlapping genetic or developmental disorders (cf., Figure 1A wherein the final result is “shared clinical phenotype”). Such inconsistencies often renders behavioral research into developmental or psychiatric disorders frustrating and such anomalous findings mask the differences that do exist. I propose that inconsistent behavioral results observed in clinical populations as well as mouse models do not infer the lack of cognitive impairments, but rather these "null" data reflect the often startling insensitivity of the behavioral tasks commonly employed.
In situations where, based on standardized behavioral tasks, mouse models do not appear to specifically model clinical phenotypes observed in patient populations, one strategy is to evaluate intermediate- or endophenotypes associated specifically with the genetic mutation and subserved by neuroanatomical structures disrupted by the mutation (cf., Figure 1B, Figure 2). A similar process applies to studies of human clinical populations when standardized tests fail to uncover phenotypes that are present, but only manifest at a subclinical level (cf., Gottesman & Gould, 2003; Hunsaker, 2012a,b; Simon, 2008, 2011).
Endophenotypes are collections of quantitative traits hypothesized to represent risk for genetic disorders at more biologically (and empirically) tractable levels than the full clinical phenotype; which often contains little more than profound deficits shared across various genetic disorders (Gould & Einat, 2007). A behavioral endophenotyping approach facilitates the identification of behavioral deficits that are clearly associated with both the specific genetic mutation and the pathological features observed in the clinical populations being modeled -- and more importantly with the pathological/clinical features unique to the population being modeled (cf., Figure 2). When designed to evaluate such disease-specific hypotheses, behavioral endophenotypes model quantitative patterns of behavioral deficits that scale with the size and / or severity of the genetic mutation (Gottesman & Gould, 2003; Gould & Gottesman, 2006; Hasler et al., 2006; Hunsaker, 2012a; Hunsaker et al., 2012; Weiser et al., 2005).
The behavioral endophenotyping process deviates from the currently accepted method for determining behavioral phenotypes. The present method is to determine phenotypes in clinical populations and mouse models -- that of using behavioral tasks that were designed without prior consideration of the pathology and anecdotal features present in the population. Far too often an approach such as this is not sufficiently sensitive to characterize gene-brain-behavior interactions that underlie disease pathogenesis (Amann et al., 2010; Gur et al., 2007; Hunsaker, 2012b; Karayiorgou et al., 2010; Simon, 2007, 2008, 2011). In contrast with the currently utilized approach, behavioral endophenotyping emphasizes the use of behavioral paradigms that were developed to specifically evaluate a priori hypotheses concerning the alterations to nominal gene-brain-behavior interactions identified or proposed to exist in a given population using carefully selected tasks to identify unique phenotypes within each model; and thus are more capable of characterizing the neurocognitive consequences of the specific gene mutations underlying the genetic disorder (Gould & Gottesman, 2006; cf., Figure 1B wherein the final step results in separate phenotypes directly related to a disease or mutation; Figure 2).
In this chapter I will evaluate advances in the methods associated with neurobehavioral endophenotyping, and will propose a clear strategy to efficiently and comprehensively characterize neurobehavioral deficits in mouse models of genetic disorders. This approach uses neurocognitive theory to design and select behavioral tasks that test specific hypotheses concerning the genetic disorder being studied. I propose this novel approach will extend the utility of mouse models by integrating the expertise of clinical neurology and cognitive neuroscience into the mouse behavioral laboratory.
Additionally, I will discuss a new collection of psychological assessments called the NIH Toolbox. The NIH toolbox was designed with the intent of extending the traditional behavioral phenotyping approach commonly used in animals into human clinical populations (cf., web resources at http://nihtoolbox.org; Gershon, 2007; Gershon et al., 2010). Although limited in application, the development of the NIH Toolbox assessments is important for research into genetic disease models since having a standardized set of experiments in human clinical research gives behavioral neuroscientists a baseline against which to develop murine behavioral paradigms.
I propose that directly emphasizing the reciprocal translation of research between human disease states and the associated mouse models is essential for both groups to mutually inform each other’s research to more efficiently generate hypotheses and elucidate treatment strategies, as has been the primary emphasis as late of the National Institutes of Health (NIH; cf., National Center for Advancing Translational Science (NCATS)). This type of translational science requires not only including the NIH Toolbox in studies of human clinical populations, but also extending beyond the NIH Toolbox to evaluate specific disease-related hypotheses that can be used as outcome measures or biomarkers for future studies into disease related risk. Similarly, researchers studying the behavior of mouse disease models also must extend beyond their comfort zones to make the scientific advancements that are required to inform progress taking place in the clinic.
Factors to Consider when Designing Behavioral (Endo)Phenotyping Batteries
Any discussion concerning the behavioral phenotyping of mouse models of genetic disorders must necessarily begin with a description of what a behavioral phenotype is and what assumptions underly tasks used to evaluate them. In short, behavioral phenotyping quantifies performance of mutant mice across behavioral experiments; and the behavioral performance is related to the clinical population to identify parallels that may exist. The analogy between the phenotype of human genetic disorder and the behavioral phenotype of the mouse model can be expressed as a combination of three factors: face validity, construct (or content) validity, and predictive validity (cf., Crawley, 2004; Guion, 1977; Hunsaker, 2012a).
Face validity is the surface similarity between the behavior of the mouse model and the patient on analogous tasks (i.e., does the performance of the mouse and human resemble each other at face value). In other words, if a mouse has to perform a similar response during a task as the patient makes during performance of a similar task, the task shows face validity. Similarly, if the mouse and human behavioral tasks can be intuitively interpreted as being similar, the task shows face validity.
Construct (or content) validity, so far as behavioral paradigms are concerned, refers to the similarity between the behavioral or cognitive domains being tested by a given task in the mouse model and the human patient. This means that for tests to show construct validity, the tasks must be designed to directly model specific aspects of the genetic disorder and that performance be subserved by similar neural substrates and / or cognitive process across species. More specifically, the tasks need to be developed to explicitly model the human disorder, not solely rely on creative post hoc interpretations of behavioral performance on general behavioral tasks (cf., Figures 1, 2; Hunsaker, 2012a,b). One necessity of construct validity is that a basic understanding of the disorder being modeled is required, such that the research translates a behavioral phenotype across species, not providing the primary elucidation of any phenotype at all in the mouse model -- although this approach can provide useful data in limited situations such as in cases of rare genetic disorders.
Predictive validity refers to the utility of a mouse model as a proxy for the patient in studies of disease progression or therapeutic intervention--this can refer to either the endpoints of a behavioral study or the physiology of the model. Although predictive validity is commonly thought of as a characteristic of phenotyping approaches, it is more accurate to state that predictive validity is the quantified endpoint of an adequately designed behavioral phenotyping experiment--that is, to define some behavior or set of behaviors that serve as valid outcome measures for later studies (Berge, 2011; Greene-Schloesser et al., 2011; Hunsaker, 2012a). In other words, predictive validity is only present when behavioral performance of the model during a given experiment proves useful for inferring or correlating dosage of a given mutation, disease progression, or treatment outcomes in not only the model, but also the clinical population.
Approaches to Endophenotyping
Since the tacit acceptance of the water maze, Barnes maze, passive/active avoidance, and contextual fear conditioning as the standard memory tasks for mouse models of disease (cf., Hunsaker, 2012a,b; Llano Lopez, et al., 2010; Whishaw & Tomie, 1996), the development of behavioral tasks to dissect the role of brain regions affected by the mutation for memory processing has stalled--at least in mice. In contrast, during this same period the research into the neural systems underlying learning and memory processes has reached a boon in rats. An effort has begun to translate these more sophisticated paradigms developed for rats into mouse disease research, with a relatively high levels of success (Hunsaker, 2012a,b; Hunsaker, et al., 2009, 2010, 2012; Nakazawa, et al., 2004; Rondi-Reig, et al., 2006).
What has remained elusive in the field of behavioral genetics is a clear theoretical rationale underlying the choice of experiments performed on each given model (i.e., water maze and Barnes maze do not test all types of spatial memory, let alone all types of “learning and memory”). To facilitate comprehensively evaluating learning and memory processes across all mouse models, it is helpful to step back and separate learning and memory into component or attributes or domains that can be evaluated in turn (cf., Hunsaker, 2012b; Hunsaker & Kesner, 2012; Kesner & Hunsaker, 2010; Kesner & Rogers, 2004; White & McDonald, 2002). In practice, this approach allows the murine researcher to evaluate brain function in a more sophisticated manner than previously possible using the standard behavioral tasks that were not developed to test any attribute or hypothesis (cf., Hunsaker, 2012a,b).
Before moving into a closer analysis of the proposed approach, it is important to mention the pitfalls with the common memory tasks used widely in mice: the water maze, Barnes maze, passive / active avoidance, and contextual fear conditioning. All of these tasks can be useful as components of a phenotyping approach, but in themselves they do not allow researchers to specifically determine the nature of impaired memory in mouse models. For all these tasks there are uncontrolled factors relating to anxiety and, more importantly, motivational confounds involving the use of negative reinforcement as the primary motivation for task performance (cf., Barkus, et al., 2010; Hunsaker 2012a,b). Furthermore, when negative reinforcement is used for motivation -- especially when using assays such as contextual fear conditioning to evaluate spatial memory -- models demonstrating disorders in affect (i.e., depression or anxiety disorders) may demonstrate "spatial" memory deficits for reasons other than de facto impairments to spatial processing (cf., Banik & Anand, 2011).
Additionally, it has been suggested on numerous occasions that the water maze may not be an appropriate task for use in mice, as mouse performance is poorer than would be predicted when compared to performance on non-water based paradigms compared to rat performance on similar tasks (Frick, Stillner & Berger-Sweeney, 2000; cf., Whishaw & Tomie, 1996) -- and dry land alternatives for the water maze have been frustratingly slow to be adopted, despite being introduced to the field as early as the 1980s (cf., Kesner, Farnsworth & DiMattia, 1989; Llano Lopez, et al., 2010).
Attributes of Memory Processing
Table 1 outlines the first consideration in developing or choosing behavioral experiments to test mouse disease models, which is to consider what type of memory needs to be tested in the mouse. Briefly, one has to consider if the disorder being studied primarily results in an episodic (event-based) memory deficit, knowledge-based memory deficits, or executive function (rule-based) deficit (Hunsaker, 2012b; Kesner & Hunsaker, 2010; Kesner & Rogers, 2004). Once the memory system being tested is determined, then the component memory domains can be identified and tested using experiments designed with each disorder and model in mind (Hunsaker, 2012a,b; Kesner & Rogers, 2004; Simon, 2007, 2008, 2011; Figure 2).
| Phase | Event-Based | Knowledge-Based | Rule-Based |
|---|---|---|---|
| Encoding | Pattern Separation | Selective Attention | Strategy Selection |
| Transient Representations | Permanent Memory Representations | ||
| Short Term Memory | Perceptual Memory | Rule Maintenance | |
| Intermediate Term Memory | |||
| Retrieval | Consolidation | Long Term Memory Rule Maintenance | |
| Pattern Completion | Retrieval Based on Flexibility and Action | Short Term Working Memory |
Table 1: Description of the processes performed by different memory systems used in the attribute theory as applicable to research using rodents
Event based memory refers to a memory system wherein information is processed online from active sensory / perceptual data and representations formed by each memory system using those sensory / perceptual data. This is the memory system that allows for trial unique responses and behavioral performance. Knowledge-based memory is often referred to as semantic memory in the human episodic memory literature. This article will use the term knowledge-based memory, because semantic memory has an implicit language component that cannot be directly modeled in rodents. Knowledge-based memory is most analogous to the reference memory system proposed by Olton, Becker, and Handelmann (1979) than to any other taxonomic system. The rule based memory system spans both the event and knowledge-based memory systems by providing a framework to guide behavioral performance. That is, the rule based memory system is the memory system that allows an individual to generate rules and motivational contexts that guide behavioral performance across all timescales and allows for generalization across paradigms and situations.
Table 2 outlines neuroanatomical substrates underlying each attribute in mice that can be consulted to guide the development or application of behavioral tasks for mouse models of disease. Importantly, although these anatomical structures have been shown to underlie the attributes as mentioned in Table 2, this description is more of a blueprint of structures that are critically involved with these processes. Stated another way, when one brain region is shown to underlie or be involved in a process, it is more likely than not that a larger network including the candidate neuroanatomical structure actually subserves the process, and that the contributions of the larger network is more poorly understood than the role for any single structure. An example of this common over-simplification is the hippocampus: hippocampus ablations result in profound deficits for spatial and temporal processing (cf., Jerman, Kesner & Hunsaker, 2006; Hunsaker et al., 2008), but removal of inputs/outputs from the entorhinal cortex and septal nuclei result often in qualitatively similar, if not identical, behavioral deficits (cf., Hunsaker, Tran & Kesner, 2008). As such, it is more correct to state that neural networks that include the hippocampus subserve spatial and temporal processing, rather than than the hippocampus in isolation subserves these memory processes.
| Attribute | Event-Based | Knowledge-Based | Rule-Based |
|---|---|---|---|
| Spatial | Hippocampus | Parietal Cortex | Infralimbic/Prelimbic, Retrosplenial Cortex |
| Temporal | Hippocampus, Basal Ganglia | Anterior Cingulate, Infralimbic/Prelimbic | Anterior Cingulate, Infralimbic/Prelimbic |
| Sensory/Perceptual | Sensory Cortices | TE2 Cortex, Perirhinal Cortex, Piriform Cortex | Infralimbic/Prelimbic |
| Response | Caudoputamen | Precentral Cortex, Cerebellum | Precentral Cortex, Cerebellum |
| Affect | Amygdala | Agranular Insula | Agranular Insula, Infralimbic/Prelimbic |
| Executive Function | Basal Ganglia | Infralimbic/Prelimbic, Parietal Cortex | Infralimbic/Prelimbic, parietal Cortex |
| Social | Unknown Network | ||
| ProtoLanguage | Unknown Network |
Table 2: Primary neuroanatomical correlates underlying each attribute in rodents
Selection and Design of Cognitive Tasks for Humans: The NIH Toolbox
The NIH Toolbox, an NIH Blueprint for Neuroscience Initiative that began in fall 2006, set out to develop a set of brief, validated instruments to assess cognitive, emotional, motor, and sensory function across a wide age range (3-85 years of age; cf., Gershon, 2007; Gershon et al., 2010). As developed, the NIH Toolbox is intended for use in epidemiological and longitudinal studies to identify those aspects of cognition that are associated with optimal function and health. similarly, the NIH Toolbox is also intended for use in large-scale intervention and prevention trials.
At present, there are many ways in which data on neurocognitive function are collected. Unfortunately, the tools currently in common use are not standardized sufficiently to allow comparison among laboratories and protocols. This results in issues similar to the situation observed in murine research as described above (cf., Wahlsten et al., 2003a). By adopting a standard set of publicly available tools, the NIH Toolbox will be able to enable the efficient aggregation of data from multiple studies and perhaps even facilitate comparison across studies. Such features as these greatly enhance the value of information collected during the course any single research project. Importantly, by providing access to the exact tools and clear instructions for usage, the site to site variability in the results from NIH Toolbox assessments can be somewhat mitigated, such that the data can be compared across sites.
The NIH Toolbox currently includes 108 primary and supplemental instruments assessing the following constructs: Cognition, emotional health, motor function, and sensory function. A full listing of paradigms is available at the NIH Toolbox web site (Gershon, 2007; Gershon et al., 2010; cf., Hoffman, Cruickshanks, & Davis, 2009; McClelland & Cameron, 2011; Pilkonis et al., 2012; Quatrano & Cruz, 2011; Wang et al., 2011) and will not be explicitly listed here, other than to point out that under each parent domain there are a number of subdomains that cover a much broader spectrum of function than the four parent domains would initially suggest. For example, the behavioral assays under the domain of Cognition include tests of attention, executive function, processing speed, working memory, episodic memory, and language subdomains. The surveys that assess the Emotional health domain evaluate positive affect, negative affect, social relationships, and stress and coping. To evaluate Motor function, the NIH Toolbox includes tests of locomotion, strength, nonvestibular balance, endurance, and dexterity. To evaluate Sensory function, the NIH Toolbox uses paradigms evaluating vision, audition, vestibular balance, taste, olfaction, and somatosensation.
The NIH Toolbox consists of these four parent domain batteries, each requiring an average of 30 min to administer (and approximately 20 min for children aged 3–5 years), with a total of 2 h administration time for the entire Toolbox (80-90 min for children). Importantly for studies involving children, each test has an upper time limit of 5-7 min, so the tests do not provide the complication of taxing the attentional capacity of children 3-5 years of age, although it is presumed adults would also appreciate the rapid nature of the tasks.
Training materials are available and test administrators do not need any specialized educational background. Instruments were developed using modern psychometric methods when possible, such as being designed for computerized adaptive testing (CAT) based on item resonance theory (IRT; Gershon, 2007; Gershon et al., 2010) and are designed to be delivered using computer workstations. These tools have undergone extensive calibration and evaluation of reliability and validity in samples ranging from 100–7500 people per instrument (total n=4705 with n=500 retested at 7 days). A national norming study commenced in early 2011, and the NIH Toolbox became publicly available in early 2012.
Utility of the NIH Toolbox in Clinical Populations
Previous to the introduction of the NIH Toolbox, behavioral and cognitive research using clinical population was hampered by similar problems as murine research: that is, different labs use similar paradigms in their research, but these tasks lack standardization. The effect of this lacking standardization is that labs are likely finding differences among their populations not because of actual differences in disease severity or observed phenotype, but rather due to small design or methodological differences in the behavioral tasks (i.e., different measures of executive function resulting in different neurocognitive phenotypes -- cf., Wisconsin Card Sort and Color / Word Stroop vs. Behavioral Dyscontrol Scale-II (BDS-II); Allen et al., 2011; Grigsby et al., 2008). This has long been an issue with mouse research and has led to the development of standardized batteries of behavioral tasks such as SHIRPA (cf., Hatcher et al., 2001; Rogers et al., 1997, 1999, 2001).
The development of the NIH Toolbox can be thought of as analogous to this movement toward a standardized set of behavioral protocols for the behavioral phenotyping of mouse disease models. Unfortunately, the movement toward standardization in mouse studies resulted in a number of tasks being accepted as standard (e.g., water maze, fear conditioning, rotarod, etc.), but did not result in standardized protocols across laboratories for these paradigms. This lack of true standardization has led to discrepant data across studies using the same model and "same" tasks, adding a level of ambiguity to any reported behavioral findings (cf., Wahlsten, 2011; Wahlsten et al., 2003a). In this way, the NIH Toolbox has improved upon the process used in mouse behavioral research by offering clear, automated, computerized protocols along with standardized, widely available apparatus for the few tools that must be purchased. The NIH Toolbox also offers a wide array of computerized analysis tools and standardized scoring means via cooperation across NIH agencies (Gershon, 2007).
Unfortunately for the traditional behavioral phenotyping strategy in mice as well as the NIH Toolbox, these batteries or collections of tools do not take into account the disease state of the mouse model or the clinical population. Using a standard set of tools rather than tools selected based on specific hypotheses does provide data that can compared against clear, accepted norms, but any interpretation of observed findings is often tortured (cf., Figure 1, 2).
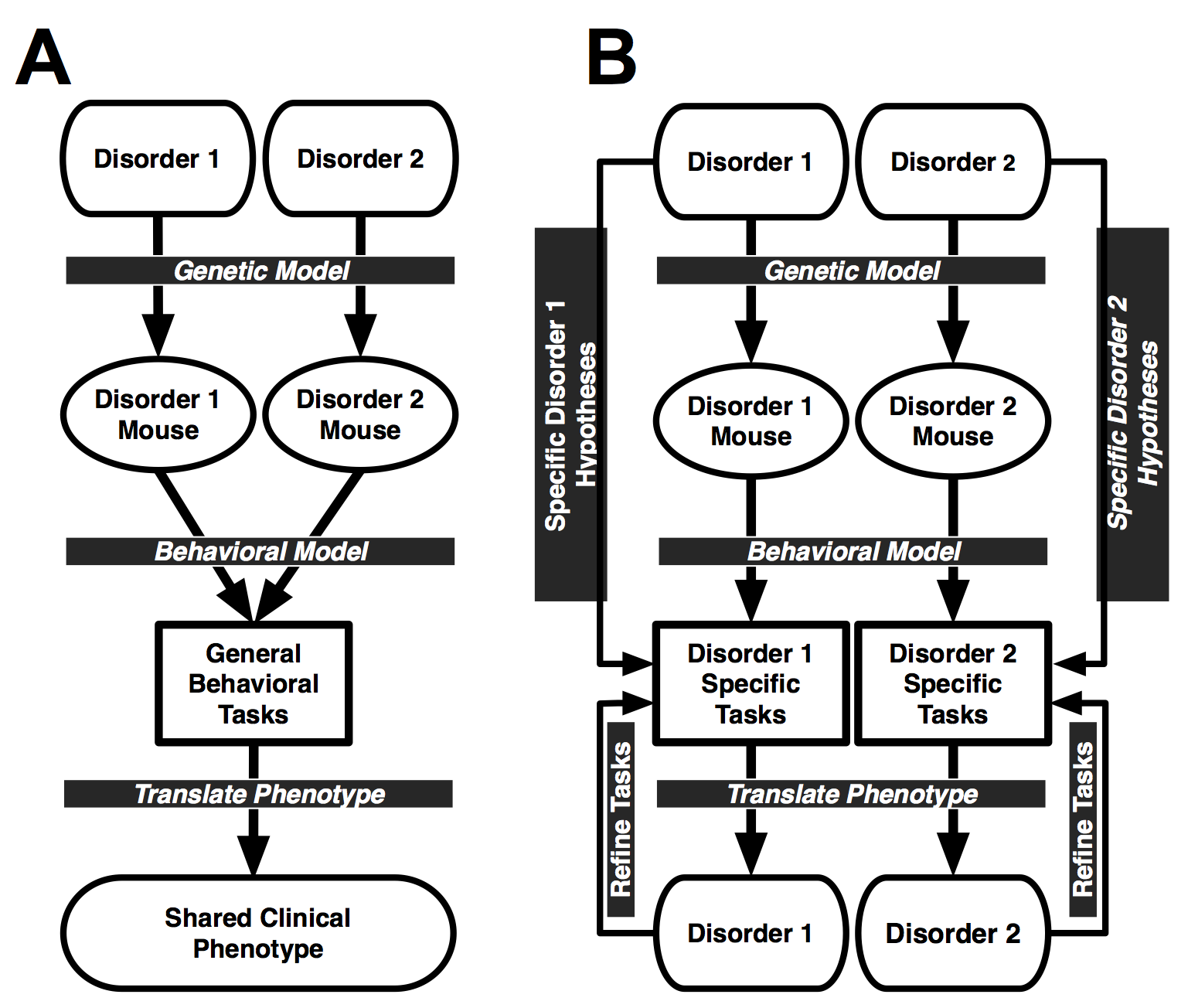 Figure 1: A. Diagram of standard behavioral phenotyping process in which different mouse models are given the same battery of tasks to define a behavioral phenotype. The outcome of the behavioral tasks are compared to the full clinical phenotype of the genetic disorders being modeled. This approach lacks the specificity and selectivity to identify phenotypes unique to a single disorder. This is a representation of the standard mouse behavioral phenotyping methods (e.g., SHIRPA) as well as the NIH Toolbox. B. Diagram of behavioral endophenotyping process in which disorder-specific hypotheses are used to develop unique batteries of behavioral tasks that directly translate to the phenotype of the clinical disorder. This approach does not model the general deficits seen across genetic disorders, but rather specifically identifies phenotypes known to be unique to the genetic disorder being modeled. Figure 2 expands upon Figure 1B. From Hunsaker, 2012a
Figure 1: A. Diagram of standard behavioral phenotyping process in which different mouse models are given the same battery of tasks to define a behavioral phenotype. The outcome of the behavioral tasks are compared to the full clinical phenotype of the genetic disorders being modeled. This approach lacks the specificity and selectivity to identify phenotypes unique to a single disorder. This is a representation of the standard mouse behavioral phenotyping methods (e.g., SHIRPA) as well as the NIH Toolbox. B. Diagram of behavioral endophenotyping process in which disorder-specific hypotheses are used to develop unique batteries of behavioral tasks that directly translate to the phenotype of the clinical disorder. This approach does not model the general deficits seen across genetic disorders, but rather specifically identifies phenotypes known to be unique to the genetic disorder being modeled. Figure 2 expands upon Figure 1B. From Hunsaker, 2012a
 Figure 2: Extension of Figure 1B with a focus on what the behavioral endophenotyping process entails. Note that all aspects of disease ranging from neuropathologic features to systemic pathology to the findings of relevant animal models are included as components that comprise the disease specific hypotheses. Differential effects of disease states on these factors provide the raw data for hypothesis generation and behavioral dissociations of diseases. Importantly, this figure emphasizes that unless two disorders show identical sequelae and pathologies, one cannot be adequately characterized using the same paradigms as those used to characterize the other. Also emphasized is the use of the full spectrum of results from the mouse model being applied to inform the hypotheses concerning the genetic/clinical disorder. This emphasis is the goal for any translational study--that the clinic and the mouse model mutually inform each other's hypotheses.
Figure 2: Extension of Figure 1B with a focus on what the behavioral endophenotyping process entails. Note that all aspects of disease ranging from neuropathologic features to systemic pathology to the findings of relevant animal models are included as components that comprise the disease specific hypotheses. Differential effects of disease states on these factors provide the raw data for hypothesis generation and behavioral dissociations of diseases. Importantly, this figure emphasizes that unless two disorders show identical sequelae and pathologies, one cannot be adequately characterized using the same paradigms as those used to characterize the other. Also emphasized is the use of the full spectrum of results from the mouse model being applied to inform the hypotheses concerning the genetic/clinical disorder. This emphasis is the goal for any translational study--that the clinic and the mouse model mutually inform each other's hypotheses.
Neglecting to take into account the specific skills and weaknesses of the population being studied results in not only misidentifying or mischaracterizing a population, but also missing behavioral or cognitive phenotypes entirely by omitting / neglecting necessary tests that would have uncovered a potential trove of information (Hunsaker 2012a; Figure 2). To provide a hypothetical example, consider a situation in which an individual with type I bipolar disorder and an individual with schizophrenia show similar patterns of performance on the NIH Toolbox collection of assessments. In this case the researcher is forced to either accept the NIH Toolbox is insufficient to characterize these populations and dissociate them from each other, or else to accept that type I bipolar disorder and schizophrenia have the same clinical presentation -- at least on the assessments being used. Clearly, type I bipolar disorder and schizophrenia do not share an overall neurocognitive phenotype, but there is no clear way to tease them apart unless clear hypotheses concerning these two disease states are used to develop tasks to characterize them at a resolution sufficient to distinguish the two disorders (process diagrammed in Figure 2). This logic can be extended to any set of disorders whose performance on the NIH Toolbox may be compared. This is where an endophenotyping strategy becomes a necessity.
That being said, it is by no means necessary or even advisable to reject the NIH Toolbox or a standardized behavioral battery outright, but it is necessary to accept the weaknesses inherent in using collections of experiments selected without regard to the population being studied. Figure 1 provides a diagrammatic representation of this argument with the NIH Toolbox / standard behavioral task battery represented by the left plate (Figure 1A) and an endophenotype / hypothesis driven approach on the right (Figure 1B). An analysis of these two approaches suggests that at best only large-scale cognitive / psychological disturbances can be identified with the NIH Toolbox that must be more carefully followed up on to provide information that can be acted upon in the development of outcome measures or therapeutic strategies. Looking at Figure 2 best illustrates how different hypotheses for different disorders emerge, namely through a consideration of all disease related sequelae and how they may affect psychological and cognitive function. Comparing the sequelae for different disorders illustrates an important point: no two disorders present with identical systemic, pathologic, endocrine dysfunction, etc. so it follows that separate disorders should be not be tested using identical tools if the goal is to uncover the differential phenotypes of unique pathological states.
Aside from being a scaffold upon which murine researchers can design behavioral paradigms, the NIH Toolbox will also prove invaluable for research into human clinical populations. As mentioned above, the NIH Toolbox allows the NIH to pool results across a number of neurological and psychiatric disorders and collect longitudinal data that can be directly compared over time. Important to achieving this goal is the fact that the NIH Toolbox was designed to be valid in populations ranging from 3-85 years of age, and the specific tools have been validated in these populations. It must be noted, however, that this comparison among laboratories and disorders in and of itself is insufficient rationale for adopting the NIH Toolbox. The researcher also would have to adopt the explicit goal of providing this initial behavioral screening to a population to be followed up on by either themselves or subsequently by other researchers to characterize and expand upon any deficits identified by the NIH Toolbox. Additionally, it is also essential that it be understood that null fundings using the NIH toolbox tools do not mean that no deficits are present in the population, just that the NIH Toolbox assessments were insufficiently sensitive to uncover any behavioral deficits. Because these tools were developed and adopted without regard to populations being studied, these tools are ill suited to characterize specific deficits or strengths in any population, but rather emphasize global deficits known to interfere with daily function.
Specifically, the NIH Toolbox has been included in proposed common outcome measures for studies into the neurologic effects of Traumatic Brain Injury (TBI). Wilde et al. (2010) evaluated a number of outcome measures, including a then under development NIH Toolbox for evaluating the neurocognitive consequences of TBI in a proposal for standard outcome measures that can be compared across institutions. Such standardization is critical for efforts toward developing therapeutics, since the choice of laboratory for validating a given intervention may be more important than the intervention used if there is no standardization among laboratories.
Similarly, the NIH Toolbox is in use for studies investigating the neurocognitive sequelae associated with epilepsy. Similarly to the case of TBI, research into epilepsy is often complicated by subtle differences among the behavioral protocols used across laboratories. Importantly, in studies of epilepsy, the use of the NIH Toolbox is as a part of a general overall health-related quality of life survey. (cf., Nowinsky et al., 2010)
Most importantly, the NIH Toolbox is intended to be brief (~2 hrs for the entire collection of assessments), minimally burdensome to respondents and administrators, relatively low in cost, psychometrically sound, free of intellectual property issues, and appropriate for use across a wide age range and with diverse populations (e.g., English and Spanish speakers). All these qualities are expected to make its use attractive to investigators.
The appeal of the NIH Toolbox is clear for those already planning to assess cognitive, emotional, motor or sensory function tested by the NIH Toolbox and, perhaps more importantly, to investigators who might not assess areas of function were it not for the availability of the NIH Toolbox. This should lead to a greater number of studies collecting standard data on more areas of function, which, since these data can be aggregated and directly compared, significantly increases the likelihood of making new discoveries, and identifying currently unknown relationships between function and health, and function and disease. In the studies mentioned above, for example, inclusion of the multidimensional NIH Toolbox in longitudinal epidemiological research could reveal new predictors or risk factors for developing symptoms after TBI or developing negative outcomes after epilepsy, as well as identify currently unsuspected long-term outcomes. This could lead to new prevention strategies as well as additional treatment targets. Similarly, using NIH Toolbox instruments when evaluating treatments could reveal a broader range of treatment effects then it is typically possible to do in a single study or a few studies. This kind of finding, in turn, may lead to development of or an adjustment to the existing therapeutic medications in clinical practice.
Critically, when the NIH Toolbox is used across laboratories, it becomes possible to evaluate intervention strategies using the same outcome measures across multiple sites simultaneously, a process not straightforward at present. Although this may be problematical in the long term as similar outcome measures may not be appropriate to compare across populations, standardized testing makes finding solutions to any complications easier than trying to harmonize data collected using different protocols across disparate studies.
While the focus of NIH Toolbox development has been for use in research studies, there has been considerable interest in directly utilizing it in the clinical arena. Several external projects are evaluating its use with clinical populations, including patients with Parkinson’s disease, TBI, stroke, and patients undergoing neurological rehabilitation for acute brain injury. The results of these studies will help inform its use in clinical settings.
Reshuffling of the NIH Toolbox by Attribute
The NIH Toolbox is organized, as mentioned above, into four component domains that can be administered in blocks so as to minimize interference among the tasks contained within each domain. An alternate method of categorizing the tests contained within the NIH Toolbox is by the component attribute tested by each task. Table 3 contains the NIH Toolbox tools, reshuffled by attribute or domain evaluated.
| Attribute | Event-Based | Knowledge-Based | Rule-Based |
|---|---|---|---|
| Spatial | Picture Sequence Memory | not addressed | not addressed |
| Temporal | Picture Sequence Working Memory, List Sorting Working Memory | List Sorting Working Memory | List Sorting Working Memory |
| Sensory/Perceptual | All Tests In Sensory Domain of NIH Toolbox | not addressed | |
| Response* | Grip Strength, Knee Extension Strength, Standing Balance, 4 Meter Gait Speed, 2 Minute Walk Endurance | 9 Hole Pegboard Dexterity | Flanker Inhibitory Control and Attention, Dimensional Change Card Sort |
| Affect | not addressed | Surveys in the Emotion Domain of the NIH Toolbox Subsections: Psychological Well Being, Negative Affect, Stress and Self Efficacy, Social Relationships | |
| Specific Cross-Domain Tasks Important for Research into Neurodevelopmental Disorders | |||
| Executive Function | Flanker Inhibitory Control and Attention, Dimensional Change Card Sort, List Sorting Working Memory, Pattern Comparison Speed, Oral Symbol Digit | ||
| Social | All Surveys Included in the Social Relationships Subsection of Emotion Domain of the NIH Toolbox | ||
| ProtoLanguage | Ray Auditory Verbal Learning , Picture Vocabulary, Oral Reading Recognition | ||
| * These motor tasks are related to the response attribute, Action-outcome and Stimulus-Response tasks more classically test the response attribute in both the event and knowledge based memory systems |
Table 3: NIH Toolbox reshuffled and organized by attribute
What becomes apparent when these tasks are categorized by attribute is that there are attribute domains that are not evaluated by the NIH Toolbox, and moreover, a number of attributes are only cursorily tested. Additionally, the design of the NIH Toolbox requires that only certain types of behavioral tasks be chosen, particularly those that may be administered in a very short timeframe, thus limiting the efficacy of the NIH Toolbox as a whole by disqualifying tests that may be more dense so far as data collection is concerned but require more time to administer. Unfortunately, it appears that these requirements have led to the selection of behavioral tasks that are not always an accepted standard for evaluating function within a given domain.
For example, to evaluate executive function and attention the NIH Toolbox uses a flanker inhibitory control task and a dimensional card sorting task. These tasks have a rich history in attentional research, but it can be argued that a go / no go task, a stop signal task, or a self ordered pointing task would dissect the executive function and attentional processes being evaluated in a more process-pure manner. In other words, using the NIH Toolbox there is no way to dissect a dysexecutive syndrome from focal attentional deficits or cognitive control processes, all of which affect behaviors differentially (cf., Grigsby et al., 2008).
Additionally, the separation of processing speed from attention seems an odd choice given that attentional processes are thought to underly performance on processing speed tasks. Additionally, the emotion domain lacks any true behavioral assays that can evaluate anhedonia, hyper vigilance, fear, anxiety, panic, or other mood state that may well be relevant to the study, but rather limits the analysis to a battery of surveys. These surveys are important, but often experimental studies into fear-, anxiety-, and panic-related behaviors may reveal more to the experimenter than the data collected in a survey, particularly responses to acute stress as well as the efficacy of an individual's coping mechanisms to stressors.
Intriguingly, the executive function domain lacks any analysis of stimulus-response or action-outcome processes that are encompassed by the response attribute. The closest the NIH Toolbox comes to evaluating these functions is the dimensional change card sort, which assesses the ability to form and switch cognitive sets. This is an important test, but more fundamental processes that may underlie cognitive set shifting remain unevaluated. Serial reaction time tasks or other similar sensory-response tasks would be able to quantify intact rule or stimulus-response processing at a more tangible level than serial set switching or other higher level cognitive processes.
The motor domain can be construed to tax the response attribute as most tests of response-based memory use motor output as a dependent measure, but this is something of a Procrustean solution as general motor function is at best only tangentially related to the response attribute. Additionally, as is the case with the tests of executive function and attention, the motor domain contains some tasks that are substandard to the questions commonly asked: For example the 9 hole pegboard was selected rather than the grooved pegboard as a test of dexterity (cf., Wang et al., 2011), resulting in the NIH Toolbox preferring a task that takes less time to administer, but at the cost of selecting a task that was not as data rich as the alternative; an alternative which takes only 2-3 more minutes to administer.
An additional, albeit necessary, weakness of the NIH Toolbox assessments is the relatively sparse amount of data collected by any given task. Whereas the common memory and executive function tests, for example, often take 15-30 min each to administer, the episodic memory test in the NIH Toolbox takes 7 minutes and has a limited number of trials. Similarly, the 9 hole pegboard uses only a single trial from each hand for data collection, rather than multiple trials as is common practice. In a similar manner, inclusion of tasks only available for children as young as 3 for adults and aged individuals (i.e., 3-85 years of age; Gershon et al., 2010) limits the utility of the NIH Toolbox by introducing the potential for ceiling performance during adulthood with non-ceiling performance at young and elderly ages, providing an inverted U function that may make analysis of longitudinal data difficult if not intractable. To remedy this potential issue, the NIH Toolbox utilizes computerized adaptive testing to accommodate performance differences across ages whenever possible, but it remains to be seen how reliable this method remains over time once truly longitudinal studies begin covering months to years within individual study participants.
Despite these limitations, the NIH Toolbox presents murine researchers with a unique opportunity to develop a standardized battery of behavioral tasks that are the direct parallel of a standardized battery of behavioral tests being used in clinical populations. The obvious implication for translational research is that the face, construct (content), and predictive validity can all be capitalized on by the mouse model -- at least so far as the NIH Toolbox assessments are concerned. An important consideration for murine behavioral researchers developing tasks to phenocopy the NIH Toolbox, there is no need to limit the task selection in a manner similar to the NIH Toolbox or to over-rely on face validity, but rather there is a need to replicate and extend upon the measures used in clinical populations
Mouse Variant of the NIH Toolbox Organized by Attribute
Currently implemented behavioral screens have the benefit of clear face validity as the implications of behavioral deficits on a task or collection of tasks are intuitively applicable in the context of the clinical phenotype, but often these tasks lack construct validity (cf., Hunsaker, 2012a,b). The behavioral endophenotyping process I am proposing emphasizes clearly defined construct validity across paradigms designed to test specific disease or mutation-related hypotheses. A starting point for the development of this test battery is the NIH Toolbox.
An optimal, comprehensive behavioral phenotyping strategy integrates common behavioral tasks as well as endophenotyping approaches performed across the lifespan. Such an approach is important because a number of genetic disorders show distinct early and late manifestations of disease that bear independent scrutiny. Often times, carriers of genetic mutations show few or at most subtle characteristics of later clinical disease early in life, but with increasing age these symptomatology emerge and the individuals receive a clinical diagnosis (Chonchaiya et al., 2009a,b; Pirogovsky et al., 2009; Rupp et al., 2009). This does not infer, however, that early in life these individuals are unaffected by the mutation; more likely the consequences of the mutation are present early in life, but require more sophisticated analyses to identify patterns of behavioral abnormalities (cf., Goodrich-Hunsaker et al., 2011a,b; Wong et al., 2012 ).
In cases of genetic disorders, it is useful to evaluate the cognitive domains that underly later clinical phenotypes early in life to determine if there are markers that can quantify or predict disease progression (Devanand et al., 2000; Pirogovsky et al., 2009; Salomonczyk et al., 2010; Yong-Kee et al., 2010). Research into a number of neurodegenerative disorders have been able to characterize subclinical endophenotypes early in the disease process that seem to predict the severity of the disease or rate of disease progression (Gilbert & Murphy, 2004a, 2004b; Karayiorgou et al., 2010; Salomonczyk et al., 2010; Xu et al., 2010; Yong-Kee et al., 2010).
The NIH Toolbox is an important tool that facilitates translational research across human and murine research. As demonstrated in Table 3, the NIH Toolbox can be organized into the attribute model to facilitate the development of analogous behavioral tasks for mice that show face and construct validity with the end goal of predictive validity. A critical aside is that the mouse behavioral paradigms do not have to be exact copies of the paradigms used by the NIH Toolbox, but rather need to assess the same fundamental cognitive processes. This assumption suggests that for a mouse model of behavioral deficits to model the human disorder only a similar pattern of behavioral deficits across tasks used in the NIH Toolbox is important, not the exact structure of any given task. In other words, it is nearly always better to err on the side of construct (content) rather than face validity when presented with the choice (cf., Hunsaker, 2012a,b).
Table 4 outlines a collection of simple tasks based on each component attribute that can be used to test cognitive dysfunction in mouse disease models and provide a functional analog to the NIH Toolbox. Aside from spatial attributes commonly tested, along with the temporal, response, social, and sensory/perceptual attributes, it is also critical to evaluate the role of affect, proto-language, and executive functioning attributes in mouse models of neurodevelopmental disorders, because these domains are often profoundly affected in these clinical populations (Hunsaker, 2012a,b; Simon, 2007, 2008, 2011).
| Attribute | Event-Based | Knowledge-Based | Rule-Based |
|---|---|---|---|
| Spatial | Metric Processing, Topological Processing, Magnitude Estimation, Delay Match To Place With Variable Interference, Biconditional Discrimination For Trial Unique Associations | Biconditional Discrimination, Delay Match To Place With Variable Cues, Declarative Sequence Learning, Cheeseboard | Covert Attention Tasks, Self Ordered Nonmatch To Sample |
| Temporal | Trace Conditioning, Temporal Ordering, Sequence Learning | Sequence Completion, Duration Discrimination | 5 Choice Serial Reaction Time, Peak Interval Timing, Time Left Task |
| Sensory/Perceptual | Delay Match To Sample With Variable Interference, Acoustic Startle, Pre-Pulse Inhibition, Psychonomic Threshold | Biconditional Discrimination | |
| Response | Ladder Walking Task, Acquisition Of Skilled Reaching, Working Memory For Motor Movements, Capellini Handling Task, Seed shelling tasks | Delay Match To Direction, Direction Discrimination, Nondeclarative Sequence | |
| learning | Reversal Learning, Probabilistic Reversal Learning, Operant Conditioning, Stop Signal Task, Serial Reversal Learning | ||
| Affect | Reward Contrast With Variable Reward Value | CLassical Conditioning, Trace Conditioning, Conditioned Preference, Anticipatory Contrast | Operant Conditioning, Gambling Task, Latent inhibition |
| Specific Cross-Domain Tasks Important for Murine Research into Neurodevelopmental Disorders | |||
| Executive Function | Contextually Cued Biconditional Discrimination, 5-Choice Serial Reaction Time, Operant Conditioning, Covert Attention Tasks, Intra-Extra Dimensional Set Shifting, Reversal Learning, Probabilistic (80/20) Reversal Learning, Serial Reversal Learning, Stop Signal Task, Gambling Task, Latent Inhibition | ||
| Social | Social Transmission of Food Preference , Social Novelty Detection | ||
| ProtoLanguage | Spectrographic Analysis of Ultrasonic Vocalizations |
Table 4: Murine options for an NIH Toolbox analog. Italicized tasks are proposed for the mouse NIH toolbox and the rest are recommended for in depth follow-up studies
An often overlooked, but critical, consideration in choosing behavioral assays is that of the neuropathology associated with any disorder being modeled (Figure 2). It seems an obvious point that one would choose behavioral paradigms that emphasize spatial (and temporal) processing to evaluate disorders with known hippocampal pathology (e.g., Alzheimer’s disease, Down’s Syndrome) and tasks emphasizing response learning in tasks showing clear basal ganglia pathology (e.g., Parkinson’s disease, Huntington’s Disease), but unfortunately this is not consistently taken into consideration in experiments using mouse models of genetic disorders (cf., Taylor, Greene & Miller, 2010; Wesson, Nixon, Levy & Wilson, 2011). Similarly, the NIH Toolbox appears to omit an explicit consideration of specific application of tasks that relate to common neuropathological features observed in aging and neurodegenerative disease. This is seen in the limited coverage of the NIH Toolbox for selectively testing the different attributes of memory (cf., Table 3).
In the case of the present analysis, the choice of behavioral paradigm used depends largely upon which behavioral tasks are being used in the NIH Toolbox in a particular disorder. Additionally, the mouse researcher can take the spirit or rationale behind the selection of a given behavioral paradigm and test the same specific process in the mouse model. The list of tasks provided in Table 4 is somewhat over-encompassing to be a direct one to one match with the NIH Toolbox, but the tasks presented test the same processes in a manner that is faithful to the intentions of the NIH Toolbox. More critically, all of the tasks listed in Table 2 have been either previously used or pharmacologically validated in murine and rodent models and thus only require a pilot project be undertaken for each lab, rather than a laborious development period for a novel task, prior to data collection.
Mouse Model of the NIH Toolbox: Behavioral Endophenotyping
Once the researcher has developed a mouse model for the NIH Toolbox, they may then extend beyond the NIH Toolbox assessments and apply an endophenotyping approach to select additional tasks that test disease and domain specific hypotheses. In other words, an explicit murine behavioral model of the NIH Toolbox can serve as a core service for all models, with follow up experiments that are unique to each disorder or model being tested (cf., Figures 1-2).
In cases of mouse models that have never been behaviorally assessed, having an explicit model of the NIH Toolbox allows for an easy translation of research findings across the human disorder (provided they use the NIH Toolbox to develop a clinical phenotype in the population) and mouse model. Once the initial phenotype is elucidated using a mouse model of the NIH Toolbox, then a more careful selection of behavioral assays can be selected based upon the behavioral findings and any analyses of pathology in the clinical population or mouse model.
The proposed analog of the NIH Toolbox is included in Table 4 as the italicized elements under each attribute and memory system. These selections have been made to be as simple as possible as well as relatively high throughput tasks. Other tasks included in each section tend to be more complicated tasks and are candidate tasks for follow up studies based upon the results of the initial screen. Also, these tasks that are intended for follow-up research rather than an initial screen are based directly upon paradigms used in cognitive research with clinical populations, so as to directly parallel the mouse model with human clinical populations (cf., Hunsaker, 2012a,b).
Examples of Behavioral Endophenotyping
Rodent Traumatic Brain Injury
Although not in mice, an example in parallel with the NIH Toolbox is the recent finding that using the metric and topological tasks, as well as the temporal ordering tasks listed in Table 4 in lieu of the water maze and/or Barnes maze demonstrated clear specificity in characterizing the behavioral deficits of experimental traumatic brain injury (TBI) caused by lateral fluid percussion in rats (Gurkoff et al., 2012). In this task, rats with TBI showed deficits for hippocampus-dependent behavioral performance requiring spatial and temporal processing, but spared parietal cortex function as well as spared cortical function related to identifying sensory/perceptual stimuli. This was a model for episodic memory deficits often demonstrated in TBI populations.
Gurkoff et al. (2012) asserted that the primary strengths of this approach in their hands was twofold: (1) the spatial and temporal ordering tasks they used had been used in human clinical populations previously, albeit under different task names (e.g., Categorical and coordinate; episodic sequence learning). They emphasized this fact as a strength as there are no clear analogs to the water maze or Barnes maze in the human TBI literature: rather episodic / general memory deficits uncovered by list learning tasks are not comparable to the rodent research, whereas research into the spatial and temporal processing have been previously done in TBI and nonTBI clinical populations. (2) The tasks Gurkoff et al. (2012) used did not require extensive training, as any deficits for executive function could result in behavioral deficits on tasks that require training due to not learning the rules of the task rather than any deficits for spatial and temporal processing per se. This is a very important consideration as TBI reliable results in altered executive function that interferes with adaptive function.
Additional benefits of this method in the TBI rat model is the relatively high throughput of the behavioral tasks as well as not requiring that rats with motor deficits swim. Additionally, as post traumatic stress disorder (PTSD) has been described in a high proportion of TBI cases, substituting paradigms that do not emphasize negative reinforcement mitigate any confounding influence of anhedonia in task performance (Hunsaker, 2012a). In selecting / designing their experiments, Gurkoff et al. (2012) applied the attribute model to select tasks that were most applicable to the population being studied (TBI) and developed hypotheses based upon the neuropathological features and clinical manifestation of TBI cases seen by their collaborators in the clinic, as well as taking into consideration the results of previous studies into their model (cf., procedure outlined in Figure 2; Table 4). This is analogous to using the NIH Toolbox to specifically assess episodic memory and executive function in individuals with TBI using more sensitive and standard measures than simple memory tests and clinical neuropsychological tools.
Mouse Model of Fragile X Premutation
For an example of this behavioral endophenotyping process in mice, research into the mouse model of the fragile X premutation, a polymorphic CGG repeat expansion on the FMR1 gene will be discussed. The fragile X premutation is associated with a late onset neurodegenerative disorder called fragile X-associated tremor ataxia/syndrome (FXTAS). FXTAS occurs in ~40% of male premutation carriers and ~16% of female premutation carriers and is associated with an intention tremor and cerebellar gait ataxia, as well as cognitive decline and executive function impairments (Hagerman & Hagerman, 2004; Jacquemont et al., 2004). Unfortunately, there are no agreed upon cognitive effects of the premutation on carriers not showing FXTAS motor signs (Allen et al., 2011; Hunter et al., 2011; Goodrich-Hunsaker et al., 2011a,b; Wong et al., 2012).
What has been demonstrated in premutation carriers is that, although not showing large-scale cognitive deficits, a number of studies identified spatial and temporal attentional deficits in female and male premutation carriers (Goodrich-Hunsaker et al., 2011a,b; Hashimoto et al., 2011; Hocking et al., 2012; Wong et al., 2012). These deficits are present despite the lack of large scale executive function deficits and gross memory disorders (Allen et al., 2011; Grigsby et al., 2008).
The analysis of behavioral deficits in the CGG KI mouse model of the fragile X premutation will emphasize the behavioral tasks included in Table 4. To evaluate whether the CGG KI mouse showed similar spatial and temporal attention problems as the premutation carriers reported by Goodrich-Hunsaker et al. (2011a,b), Hocking et al. (2012), and Wong et al. (2012), we applied the rationale diagrammed in Figure 2. In other words, the cognitive, pathological, and neuroendocrine phenotypes of the premutation were considered in the task design for the CGG KI mouse model.
So far as neurobehavioral deficits in the CGG KI mouse are concerned, the CGG KI mouse shows a number of basic processing deficits for spatial and temporal information. The CGG KI mouse model of the fragile X premutation shows spatial memory deficits on the water maze when they are older than 52 weeks of age (van Dam et al., 2005). These deficits, however, appear to be very mild and are not as profound as the general memory deficits demonstrated in FXTAS patients (cf., Hagerman & Hagerman, 2004; Jacquemont et al., 2004). Based on reports that suggest increasing CGG repeat lengths affect spatial attention (cf., Goodrich-Hunsaker et al., 2011a,b; Hocking et al., 2012; Wong et al., 2012), spatiotemporal function was specifically assayed in the CGG KI mice.
Using a pair of behavioral tasks to evaluate the resolution of spatial memory in CGG KI mice, it was demonstrated that CGG KI mice show deficits for mentally comparing the the specific distances that separate two objects in space. This was evaluated using a metric change detection task wherein mice are habituated to two objects on a tabletop separated by 45 cm for 15 min. After being removed from the table for 5 min, mice were returned to the tabletop with the objects placed at 30 cm separation. The ability of mice to notice a change in the distance between the objects required the mouse to remember the original distance and compare it with the current sensory input. Deficits for spatial attention tested by the metric task were present as early as 3 months of age, but in cross sectional studies these deficits did not appear to become increasingly profound in mice that were 6, 9, or 12 months of age (Hunsaker et al., 2009, 2012).
However, performance of CGG KI mice on a task that required the mice to remember which side of the tabletop was occupied by an object after the objects were transposed was not impaired at early ages. In fact, CGG KI mice did not show deficits for this topological change detection task until they were 9 and 12 months of age, with their performance not differing from wildtype littermate controls at 3 and 6 months of age (Hunsaker et al., 2009, 2012). This tasks also requires spatial attention, but of a different type than the metric task. The topological task required the mouse to remember an object-place relationship that did not require the fine-scale spatial attention required by the metric task.
What can be learned from these data are twofold: (1) that the resolution of spatial attention in CGG KI mice is profoundly reduced from a very young age compared to wildtype littermates, presumably affected relatively early during development, and this resolution appears to be fixed across time, such that the resolution does not deteriorate / progressively worsen as a function of age. (2) Performance of CGG KI mice on spatial memory tasks that do not require fine spatial attention such as the topological change detection task is not impaired at an early age, but these mice do show a progressive worsening of spatial memory across age, with deficits emerging in middle life and worsening at advanced ages in CGG KI mice, a pattern similar to those seen with the water maze (Hunsaker et al., 2009, 2012; van Dam et al., 2005).
An easily overlooked element in this pattern of deficits in the CGG KI mouse model is the dissociation between the developmental course of deficits present across the metric and topological tasks. The finding that spatial attention deficits evaluated by the metric processing task are present at a young age and do not worsen across the lifespan suggests a fundamental developmental alteration that renders the CGG KI mouse unable to overcome the interference between the distances between spatial locations before and after the metric change (i.e., the metric change was not profound enough for the mouse to discriminate the new distance between objects from the remembered object distance). CGG KI mouse performance on the topological task, however, showed a somewhat degenerative pattern. Performance of the topological task was intact in young animals, suggesting that spatial memory per se was not disrupted in the CGG KI mice. As the mice age, however, deficits for this task emerged, suggesting some effect of age and the premutation compounding to result in general spatial memory impairments. This dissociation in task performance is important because it suggests that the premutation results in reduced resolution of spatial attention, not general spatial memory deficits (i.e., the mice can identify changes in object-place associations, but lack the ability to perform comparisons between an observed distance between objects and one retrieved from memory).
To evaluate temporal memory in CGG KI mice, a temporal ordering for visual objects task was used. In this task, in a clear box mice were presented with two copies of an object for 5 min, then removed from the box for 5 min. The mice were then presented with two copes of a second object in the box for 5 min. After another 5 min break, they were exposed to two copies of a third object for 5 min. After the mice were removed after this third object exposure for a 5 min break, they received one of two tests. The first test is a temporal ordering test during which the mouse is presented with a copy of the first and a copy of the third object and allowed to explore. Typically, mice will preferentially explore the first over the third object. It has been suggested that this paradigm requires sequential learning and fine-scale temporal attention for the mouse to remember the order the stimuli were experienced and to later compare the relative order of these memories to guide behavioral choice.
On another day after a difference set of object presentation, the mice receive a second test, a novelty detection task. In this task, the first object they were presented that day as well as a never before seen novel object were presented. In general, mice will preferentially explore the novel object over the familiar one. Intact performance during this novelty task suggest that any deficits on the temporal ordering task are not due to an inability to discriminate the stimuli, general memory deficits, or forgetting the first object before the test session, as they can discriminate a familiar object from a novel object, suggesting intact visual object memory.
On these tasks, the CGG KI mice showed intact performance for the novelty detection task, but profound impairments for temporal attention as assessed by the temporal ordering task (Hunsaker et al., 2010, 2012). Again, these data suggest that the CGG KI mouse has intact sensory/perceptual processing and intact overall memory but impaired temporal attention that results in temporal ordering deficits.
As a follow up to these experiments, an explicit spatiotemporal processing task was performed based on spatiotemporal working memory tasks used in human populations (Borthwell et al., 2012; cf., Kesner, Hopkins, & Fineman, 1994). In this task, mice were presented with a large object in a first spatial location for 5 min in a large box with prominent visual cues present. After a 5 min break, the mice explored the same object in a second location. This was repeated for a third location. In this way, the mouse explored the same object in three locations, which we will call exploration of a location. After these presentations, one of three tests were given (over three days with new object-location pairings each day).
The first is a temporal ordering for spatial locations test wherein the first and third locations were marked with identical objects identical to that used to present the locations. Importantly, these locations were always 180º from the mouse's starting location, thus minimizing spatial interference between the remembered spatial locations to allow for an analysis of temporal attention for spatial location information. Preferential exploration the first over the third location was used to index spatiotemporal attention.
The second test was a pure spatial memory control during which the first location and a novel fourth location were marked by identical objects, which were 180º from the mouse's starting posits. This again minimized any crowding between the remembered spatial location and a novel spatial location. Preferential exploration do the novel location suggests intact general memory processing.
The final test was a spatial resolution test during which the first and novel object were only separated by 45-90º from the mouse's starting position, increasing the spatial interference to isolate the ability of the CGG KI mice to overcome the interference between the remembered location and a novel spatial location.
On these tasks, the CGG KI mice showed no deficit for spatiotemporal novelty detection when the locations were separated by 180º and thus spatial interference was minimized because there was no temporal or spatial interference among remembered spatiotemporal memory and a novel location. However, the CGG KI mice did show impairments when the spatial interference was maximized by placing the novel spatial location very close to a remembered spatial location. The CGG KI mice also showed temporal attention deficits for spatial information during the temporal ordering test -- strongly suggesting an inability to overcome spatial and temporal memory interference and providing clear evidence for impaired spatiotemporal attention (Borthwell et al., 2012).
An important element to these behavioral results is that both male and female CGG KI mice showed deficits. This is not a minor point as female fXPCs show reduced disease severity due to the protective effect of a second, non mutated FMR1 gene on the second X chromosome, which males lack (cf., Jacquemont et al., 2004; Schluter et al., 2012; Tassone et al., 2012). Finding these deficits in both male and female CGG KI mice suggests cognitive deficits within the domain of spatiotemporal attention are fundamental consequences of the premutation, since these deficits are present and identifiable even in the least affected subgroup within the fXPC population (cf., logic provided by Goodrich-Hunsaker, 2011a,b).
As stated above, FXTAS patients often present in the clinic with an intention tremor and/or a cerebellar gait ataxia. Importantly, the tremor and ataxia seem to present with an oscillatory component, such that the gait ataxia becomes more profound as the individual walks until they lose balance. They appear relatively normal for the first few steps, but then a postural away emerges that grows in amplitude with each step until the patient either braces against a wall or falls over. For the intention tremor, FXTAS patients appear to show normal motor function at first, but as the trial continues (i.e., spiral or Archimedes or drawing a third line in the space separating two lines), a minute oscillation emerges which increases amplitude until the patient stops. Gait ataxia shows a similar tendency with the amplitude of the postural sway increasing with each step until the patient braces themselves with a cane or against a wall, after which the pattern of increasing instability repeats (RJ Hagerman & DA Hall, unpublished observations). These data suggest there may be some sort of abnormal feedback among cortical and cerebellar systems that prevents the fine online correction of movements so errors accumulate and exacerbate out of control. In other words, it is possible the cerebellum never receives the vestibular / kinesthetic feedback that signals the accumulating error present during each movement, so the amplitude of the error term sums exponentially with each subsequent movement until the patient loses control and has to completely stop the movement to reset. The implication for these data is that tasks requiring temporally extended performance of motor movements (e.g., long trials) and/or be sufficiently difficult to induce stress may be required to induce an intention tremor or ataxic gait in any mouse model for the fragile X premutation and FXTAS.
Although this hypothesis remains untested in fXPCs, the CGG KI mouse model does in fact show the visuospatial/visuomotor deficits predicted by the above model. To specifically evaluate visuomotor functioning CGG KI mice, a skilled forelimb reaching task was developed. In this task, the mouse was required to reach through a narrow window to obtain a reward pellet just out of reach of the tongue at a 30º angle from the edge of the window (this required the mouse to reach with the non-preferred paw). The number of pellets the mouse was able to obtain without dropping or knocking away the pellet was recorded, as was the number of errors. The CGG KI mice showed a different learning curve than wildtype mice, with CGG KI mice learning the task on average 1-2 days later than wildtype litter mates and never quite learning the task to the same level of asymptotic performance (Diep et al, 2012). Importantly, these deficits were subtle, only becoming apparent when the mice were forced to perform a rather difficult task. These data suggest there is a fundamental impairment in one of two neural systems: (1) the parietal cortex and its interactions with the superior colliculus and cerebellum were unable to provide adequate spatiotemporal updating to allow the CGG KI mice to reach the same level of success as the wildtype mice. (2) the pontocerebellar system shows disruptions (as has been suggested in FXTAS) in a way we could not identify histologically and the deficits arise from an inability of the cerebellum to control the fine motor skills required to skillfully reach, grasp, and consume the reward.
The qualitative data suggests that the CGG KI mice reached with more of a circular or radial motion rather than a directed vector toward the reward pellet, and that mice with longer CGG repeat lengths showed less directed / more radial trajectories than wildtype littermate mice. This resulted in the CGG KI mice knocking the reward away or reaching through the reward, rather than showing difficulty in the grasping of the reward. Once the CGG KI mice grasped the reward pellet, however, they were able to consume it, not showing any difference in the ability to hold onto the pellet and consume it.
To evaluate potential subclinical gait ataxia or general clumsiness in the CGG KI mice, a skilled ladder walking task was employed. The apparatus developed to perform these experiments was a manual ladder rung task (Hunsaker et al., 2011b). The apparatus consisted of clear plexiglass walls separated by approximately 5 cm with 2 mm diameter steel rungs making up the floor of the apparatus. For this initial study, the mice were placed at one end of the apparatus and were allowed to walk back and forth for 2 min. The number of foot slips we recorded for the duration of the 2 min, except for when the mouse was turning around. The number of times the mouse went from one end of the apparatus to the other was also recorded as a general locomotor measure. On this task, mice as young as 2 months of age already showed an increased number of foot slips than wildtype littermate controls. Importantly, the mice showed both forelimb and hindlimb slips, something that suggests concurrent visuospatial and basic motor deficits. These data indicate the presence of visuospatial processing deficits in that there were a high number of forelimb slips in the CGG KI mice, suggesting a difficulty in planning where in space to place the forepaw as well as a difficulty for updating the movement as the step progressed (i.e., as the mouse moved forward the initial planned step has to be modified as the visual space and intended movement interact to guide correct foot placement and an inability to do so results in a foot slip). Hind foot slips however, do not have a visuospatial planning component, but rather reflect a dysfunction in motor function, albeit a subtle one. An inability, or at least increased difficulty with the hind limb placement may reflect some form of ataxia that has not been picked up using other apparatus and methods. Additionally, as a model of FXTAS, during performance of this task, the CGG KI mice showed a high frequency, low amplitude shaking behavior that was visually similar to the description of intention tremors in human FXTAS. This is important as this task was rather difficult, and may have required a high degree of effort from the CGG KI mice that was not required from the wildtype mice, that in no cases did wildtype mice ever present similar tremoring or shaking behaviors.
Now it can be clearly seen that by using a series of more specific tasks than those commonly used to behaviorally phenotype mice, a clear behavioral phenotype emerges in the CGG KI mouse (Borthwell et al., 2012; Diep et al., 2012; Hunsaker et al., 2009, 2010, 2011b, 2012; van Dam et al., 2005). More importantly, however, was the fact that the mouse behavioral phenotype phenocopies results in human premutation carriers without FXTAS (cf., Wong et al., 2012). The initial cognitive deficits were followed up by an analysis of subclinical motor deficits that may correlate with subclinical apraxia mentioned on numerous occasions by a collaborator working with the clinical FXTAS population (RJ Hagerman, personal communication).
These data suggest the CGG KI mouse is an appropriate model for the cognitive deficits present in the fragile X premutation -- at least so far as a mouse can serve as a proxy for human cognitive function. Importantly, the CGG KI mouse shows the same neuropathological features as well as systemic organ pathology present in carriers of the fragile X premutation, proving the CGG KI mouse is a valid model for the neurologic and pathological consequences of the premutation (i.e., the mouse itself shows construct validity with the fragile X premutation; Greco et al. 2006; Hunsaker et al., 2011a; Schluter et al., 2012; Tassone et al., 2012; Wenzel et al., 2010).
Although it may at first glance seem unimportant that the mouse model phenocopies the human disorder, it is critical to understand that only by directly phenocopying the human disorder can a mouse be truly used for translational research (i.e., demonstrate predictive validity; Hunsaker, 2012a). Once a mouse model is shown to recapitulate the pattern of deficits observed in the clinical population, the behavioral results in the mouse can be used as biomarkers or targets for treatment studies or experimental risk prodrome for studies into gene x environment interactions as related to the incomplete penetrance of FXTAS among premutation carriers.
Conclusions
In recent years, there has been impetus placed on developing behavioral biomarkers that can be used to predict not only later disease onset or progression, but perhaps disease severity. These collections of intermediate or behavioral endophenotypes serve as outcome measures for pharmacological interventions (Cannon & Keller, 2006; Gottesman & Gould, 2003; Gould & Gottesman, 2006; Gur et al., 2007; Hunsaker, 2012a,b). This search for behavioral biomarkers, however, has not consistently been extended into the mouse models of genetic disorders. To date, the closest research into mouse disease models comes to developing behavioral biomarkers is to thoroughly parameterize a single task and apply the biomarker as a single screen for various mouse models to choose candidates for drug studies (e.g., attenuated PPI response or audiogenic seizures for the Fmr1 KO and 22q11.2 deletion syndrome mouse models; cf., Long et al., 2006; Paylor & Lindsay 2006). The strength of the standard approach is the ability to define a canon against which to gauge later models; however, the limitation of this approach is that it lacks the ability to evaluate complimentary models of a given disease to get at the fundamental processes disrupted in the human mutation.
This limitation occurs because a model may fail to model one phenotype, even though the mouse may model any number of other phenotypes that are not included in the standard behavioral screen. This lack of sensitivity is a major limitation as studies into the therapeutic effects of pharmacological agents will be incomplete in the absence of predefined behavioral biomarkers as outcome measures. The tools available in the NIH Toolbox will hopefully alleviate a number of these issues by expanding the number of commonly used, clinically reliable tests that can be modeled in the mouse. This not only increases the amount of clinical data that can be reliably accumulated in the human disease populations, but also expands the number of potential behavioral phenotypes to be tested in the mouse model. This reciprocal dialogue among levels of research should facilitate the usefulness of mouse disease models as has never been previously possible (cf., Hunsaker, 2012a,b; Figures 1-2).
If the recent advances in the cognitive neuroscience of neurodevelopmental disorders are extended to their respective mouse models, perhaps the associated behavioral biomarkers of such disorders may not only be complimented by, but extended though use of mouse models studying the component processes underlying disease states. These well defined behavioral biomarkers can be used as correlates or covariates with molecular studies of underlying disease mechanisms in mice that cannot be directly studied in human patient populations.
References
Allen, E. G., Hunter, J. E., Rusin, M., Juncos, J., Novak, G., Hamilton, D., ... Sherman, S. L. (2011). Neuropsychological findings from older premutation carrier males and their noncarrier siblings from families with fragile X syndrome. Neuropsychology 25(3), 404-411.
Amann, L. C., Gandal, M. J., Halene, T. B., Ehrlichman, R. S., White, S. L., McCarren, H. S. & Siegel, S. J. (2010). Mouse behavioral endophenotypes for schizophrenia. Brain Res Bull 83(3-4), 147-161.
Baker, K. B., Wray, S. P., Ritter, R., Mason, S., Lanthorn, T. H. & Savelieva, K. V. (2010). Male and female Fmr1 knockout mice on C57 albino background exhibit spatial learning and memory impairments. Genes Brain Behav 9(6), 562-574.
Banik, A. & Anand, A. (2011). Loss of learning in mice when exposed to rat odor: a water maze study. Behavioural Brain Research. 216(1), 466-71.
Barkus, C., McHugh, S. B., Sprengel, R., Seeburg, P. H., Rawlins, J. N. & Bannerman, D. M. (2010). Hippocampal NMDA receptors and anxiety: at the interface between cognition and emotion. European Journal of Pharmacology, 626(1), 49-56.
Berge, O. G. (2011). Predictive validity of behavioral animal models for chronic pain. B J Pharmacology 164(4), 1195-1206.
Bohlen, M., Cameron, A., Metten, P., Crabbe, J. C. & Wahlsten, D. (2009). Calibration of rotational acceleration for the rotarod test of rodent motor coordination. J Neurosci Methods 178, 10-14.
Borthwell, R. M., Hunsaker, M. R., Willemsen, R., Berman, R. F. (2012). Spatiotemporal processing deficits in female CGG KI mice modeling the fragile X premutation. Behavioural Brain Research, 233(1), 29-34.
Cannon, T. D. & Keller, M. C. (2006). Endophenotypes in the genetic analyses of mental disorders. Annu Rev Clin Psychol 2, 267-290.
Chonchaiya, W., Schneider, A. & Hagerman, R. J. (2009a). Fragile X: a family of disorders. Adv Pediatr 56, 165-186.
Chonchaiya, W., Utari, A., Pereira, G. M., Tassone, F., Hessl, D. & Hagerman, R. J. (2009b). Broad clinical involvement in a family affected by the fragile X premutation. J Dev Behav Pediatrics 30, 544-551.
Crabbe, J.C. & Wahlsten, D. (2003). Of mice and their environments. Science 299, 1313-1314.
Crabbe, J. C., Wahlsten, D. & Dudek, B.C., (1999). Genetics of mouse behavior: interactions with laboratory environment. Science 284, 1670-1672.
Crawley, J. N. (2004). Designing mouse behavioral tasks relevant to autistic-like behaviors. Ment Ret Dev Disabil Rev 10, 248-258.
Crawley, J. N. (2007). Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol 17, 448-459.
Devanand, D. P., Michaels-Marston, K. S., Liu, X., Pelton, G. H., Padilla, M., Marder, K., ... Mayeux, R. (2000). Olfactory deficits in patients with mild cognitive impairment predict Alzheimer's disease at follow-up. Am J Psychiatry 157, 1399-1405.
Diep, A. A., Hunsaker, M. R., Kwock, R., Kim, K. M., Willemsen, R., & Berman, R. F. (2012). Female CGG knock-in mice modeling the fragile X premutation are impaired on a skilled forelimb reaching task. Neurobiology of Learning and Memory, 97, 229-234.
Farley, S. J., McKay, B. M., Disterhoft, J. F. & Weiss C. (2011). Reevaluating hippocampus- dependent learning in FVB/N mice. Behavioral Neuroscience, 125(6), 871-8.
Frick, K. M., Stillner, E. T. & Berger-Sweeney, J. (2000). Mice are not little rats: species differences in a one-day water maze task. Neuroreport, 11(16), 3461-5.
Gershon, R. C. (2007). NIH toolbox: Assessment of neurological and behavioral function. NIH (contract HHS-N-260-2006 00007-c) http://www.nihtoolbox.org.
Gershon, R. C., Cella, D., Fox, N. A., Havlik, R. J., Hendrie, H. C. & Wagster, M. V. (2010). Assessment of neurological and behavioural function: the NIH Toolbox. Lancet Neurol. 9(2):138-139.
Gilbert, P. E. & Murphy, C. (2004a). Differences between recognition memory and remote memory for olfactory and visual stimuli in nondemented elderly individuals genetically at risk for Alzheimer's disease. Exp Gerontol 39, 433-441.
Gilbert, P. E. & Murphy, C. (2004b). The effect of the ApoE epsilon4 allele on recognition memory for olfactory and visual stimuli in patients with pathologically confirmed Alzheimer's disease, probable Alzheimer's disease & healthy elderly controls. J Clin Exp Neuropsychol 26, 779-794.
Goodrich-Hunsaker, N. J., Wong, L. M., McLennan, Y., Srivastava, S., Tassone, F., Harvey, D., ... Simon, T. J.(2011a). Young adult female fragile X premutation carriers show age- and genetically-modulated cognitive impairments. Brain Cogn 75, 255-260.
Goodrich-Hunsaker, N. J., Wong, L. M., McLennan, Y., Tassone, F., Harvey, D., Rivera, S. M. & Simon, T. J. (2011b). Adult Female Fragile X Premutation Carriers Exhibit Age- and CGG Repeat Length-Related Impairments on an Attentionally Based Enumeration Task. Front Hum Neurosci 5, 63.
Gottesman, I. I. & Gould, T. D. (2003). The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry 160, 636-645.
Gould, T. D. & Einat, H. (2007). Animal models of bipolar disorder and mood stabilizer efficacy: a critical need for improvement. Neurosci Biobehav Rev 31, 825-831.
Gould, T. D. & Gottesman, I. I. (2006). Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav 5, 113-119.
Greco, C. M., Berman, R. F., Martin, R. M., Tassone, F., Schwartz, P. H., Chang, A., ... Hagerman, P. J. (2006). Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain. 129(Pt 1),243-55.
Greene-Schloesser, D. M., Van der Zee, E. A., Sheppard, D. K., Castillo, M. R., Gregg, K. A., ... Bult-Ito, A. (2011). Predictive validity of a non-induced mouse model of compulsive-like behavior. Behav Brain Res 221, 55-62.
Grigsby, J., Brega, A. G., Engle, K., Leehey, M. A., Hagerman, R. J., Tassone, F., ... Reynolds, A. (2008). Cognitive profile of fragile X premutation carriers with and without fragile X-associated tremor/ataxia syndrome. Neuropsychology 22(1), 48–60.
Guion, R. M. (1977). Content Validity--The Source of My Discontent. App Psychol Meas 1, 1-10.
Gur, R. E., Calkins, M. E., Gur, R. C., Horan, W. P., Nuechterlein, K. H., Seidman, L. J. & Stone, W. S. (2007). The Consortium on the Genetics of Schizophrenia: neurocognitive endophenotypes. Schiz Bull 33, 49-68.
Gurkoff, G. G., Gahan, J. D., Ghiasvand, R. T., Hunsaker, M. R., Feng, J. F., Berman, R. F., ... Folkerts, M. M. (2012). Moderate lateral fluid percussion injury results in in the metric and temporal ordering but not topological working memory tasks. Journal of Neurotrauma.
Hagerman, P. J., & Hagerman, R. J. (2004). The fragile-X premutation: a maturing perspective.Am J Hum Genet. 74(5), 805-16.
Hasler, G., Drevets, W. C., Gould, T. D., Gottesman, I. I. & Manji, H. K. (2006). Toward constructing an endophenotype strategy for bipolar disorders. Biol Psych 60, 93-105.
Hatcher, J. P., Jones, D. N., Rogers, D. C., Hatcher, P. D., Reavill, C., Hagan, J. J. & Hunter, A. J. (2001). Development of SHIRPA to characterise the phenotype of gene-targeted mice. Behav Brain Res 125, 43-47.
Hocking, D. R., Kogan, C. S., & Cornish, K. M. (2012). Selective spatial processing deficits in an at-risk subgroup of the fragile X premutation. Brain Cogn. 79(1):39-44.
Hoffman, H. J., Cruickshanks, K. J., & Davis, B. (2009). Perspectives on population-based epidemiological studies of olfactory and taste impairment. Ann N Y Acad Sci 1170:514-30.
Hunsaker, M. R. (2012a). Comprehensive neurocognitive endophenotyping strategies for mouse models of genetic disorders. Prog Neurobiol 96(2), 220-241.
Hunsaker, M. R. (2012b). The importance of considering all attributes of memory in behavioral endophenotyping of mouse models of genetic disease. Behav Neurosci 2012 Jun;126(3):371-80.
Hunsaker, M. R., Fieldsted, P. M., Rosenberg, J. S. & Kesner, R. P. (2008). Dissociating the roles of dorsal and ventral CA1 for the temporal processing of spatial locations, visual objects, and odors. Behavioral Neuroscience, 122(3), 643-50.
Hunsaker, M. R., Goodrich-Hunsaker, N. J., Willemsen, R., & Berman, R. F. (2010). Temporal ordering deficits in female CGG KI mice heterozygous for the fragile X premutation. Behavioral Brain Research, 213(2), 263-268.
Hunsaker, M. R., Greco, C. M., Spath, M. A., Smits, A. P. T., Navarro, C. S., Tassone, F., … Hukema, R. K. (2011a). Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathologica. 122(4), 467-479.
Hunsaker, M. R., Kim, K. M., Willemsen, R., Berman, R. F. (2012). CGG Trinucleotide repeat length modulates neural plasticity and spatiotemporal processing in a mouse model of the fragile X premutation. Hippocampus.
Hunsaker, M. R., Tran, G. T. & Kesner, R.P. (2008). A double dissociation of subcortical hippocampal efferents for encoding and consolidation/retrieval of spatial information. Hippocampus, 18, 699-709.
Hunsaker, M. R., von Leden, R. E., Ta, B. T, Goodrich-Hunsaker, N. J., Arque, G., Kim, K. M., … Berman, R. F. (2011b). Motor deficits on a ladder rung task in male and female adolescent CGG knock-in mice. Behavioural Brain Research, 222, 117-121.
Hunsaker, M. R., Wenzel, H. J., Willemsen, R., & Berman, R. F. (2009). Progressive spatial processing deficits in a mouse model of the fragile X premutation. Behavioral Neuroscience. 123(6), 1315-1324.
Jacquemont, S., Hagerman, R. J., Leehey, M. A., Hall, D. A., Levine, R. A., Brunberg, J. A., ... Hagerman, P. J. (2004). Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 291(4), 460-9.
Jerman, T. S., Kesner R.P. & Hunsaker, M. R. (2006). Disconnection analysis of CA3 and DG in mediating encoding but not retrieval in a spatial maze learning task. Learning and Memory, 13(4), 458-464.
Karayiorgou, M., Simon, T. J. & Gogos, J. A. (2010). 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nat Rev Neurosci 11, 402-416.
Kendler, K. S. & Neale, M. C. (2010). Endophenotype: a conceptual analysis. Mol Psychiatry 15, 789-797.
Kesner, R. P., Farnsworth, G., DiMattia, B. V. (1989). Double dissociation of egocentric and allocentric space following medial prefrontal and parietal cortex lesions in the rat. Behavioral Neuroscience, 103(5), 956-961.
Kesner, R. P., Hopkins, R. O., & Fineman, B. (1994). Item and order dissociation in humans with prefrontal cortex damage. Neuropsychologia. 32(8), 881-91.
Kesner, R. P. & Hunsaker, M. R. (2010). The temporal attributes of episodic memory. Behavioural Brain Research, 215(2), 299-309.
Kesner, R. P. & Rogers, J. R. (2004). An analysis of independence and interactions of brain substrates that subserve multiple attributes, memory systems & underlying processes. Neurobiology of Learning and Memory, 82(3), 199-215.
Long, J. M., LaPorte, P., Merscher, S., Funke, B., Saint-Jore, B., Puech, A., ... Wynshaw-Boris, A., (2006). Behavior of mice with mutations in the conserved region deleted in velocardiofacial/DiGeorge syndrome. Neurogenetics 7, 247-257.
Llano Lopez, L., Hauser, J., Feldon, J., Gargiulo, P. A. & Yee, B. K. (2010). Evaluating spatial memory function in mice: a within-subjects comparison between the water maze test and its adaptation to dry land. Behav Brain Res 209, 85-92.
Manji, H. K., Gottesman, I. I. & Gould, T. D. (2003). Signal transduction and genes-to-behaviors pathways in psychiatric diseases. Sci STKE 2003, pe49.
McClelland, M. M., & Cameron, C. E. (2011). Self-regulation and academic achievement in elementary school children. In R. M. Lerner, J. V. Lerner, E. P. Bowers, S. Lewin-Bizan, S. Gestsdottir, & J. B. Urban (Eds.), Thriving in childhood and adolescence: The role of self-regulation processes. New Directions for Child and Adolescent Development, 133, 29–44.
Nakazawa, K., McHugh, T. J., Wilson, M. A. & Tonegawa, S. (2004). NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci 5, 361-372.
Nakazawa, K., Sun, L. D., Quirk, M. C., Rondi-Reig, L., Wilson, M. A. & Tonegawa, S. (2003). Hippocampal CA3 NMDA receptors are crucial for memory acquisition of one-time experience. Neuron 38, 305-315.
Nowinski, C. J., Victorson, D., Cavazos, J. E., Gershon, R., & Cella, D. (2010). Neuro-QOL and the NIH Toolbox: implications for epilepsy. Therapy. 7(5):533-540.
Olton, D. S., Becker, J. T. & Handelmann, G. E. (1979). Hippocampus, space & memory. Brain and Behavioral Science, 2, 313-365.
Paylor, R. & Lindsay, E., (2006). Mouse models of 22q11 deletion syndrome. Biol Psychiatry 59, 1172-1179.
Pilkonis, P. A., Choi, S. W., Salsman, J. M., Butt, Z., Moore, T. L., Lawrence, S. M., ... Cella, D. (2012). Assessment of self-reported negative affect in the NIH Toolbox. Psychiatry Res.
Pirogovsky, E., Goldstein, J., Peavy, G., Jacobson, M. W., Corey-Bloom, J. & Gilbert, P. E. (2009) Temporal order memory deficits prior to clinical diagnosis in Huntington's disease. J Int Neuroopsych Soc 15, 662-670.
Quatrano, L. A., & Cruz, T. H. (2011). Future of outcomes measurement: impact on research in medical rehabilitation and neurologic populations. Arch Phys Med Rehabil. 92(10 Suppl):S7-11.
Rogers, D. C., Fisher, E. M., Brown, S. D., Peters, J., Hunter, A. J. & Martin, J. E. (1997). Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome 8, 711-713.
Rogers, D. C., Jones, D. N., Nelson, P. R., Jones, C. M., Quilter, C. A., Robinson, T. L. & Hagan, J. J. (1999). Use of SHIRPA and discriminant analysis to characterise marked differences in the behavioural phenotype of six inbred mouse strains. Behav Brain Res 105, 207-217.
Rogers, D. C., Peters, J., Martin, J. E., Ball, S., Nicholson, S. J., Witherden, A. S., ... Fisher, E. M. (2001). SHIRPA, a protocol for behavioral assessment: validation for longitudinal study of neurological dysfunction in mice. Neurosci Lett 306, 89-92.
Rondi-Reig, L., Petit, G. H., Tobin, C., Tonegawa, S., Mariani, J. & Berthoz, A. (2006). Impaired sequential egocentric and allocentric memories in forebrain-specific NMDA receptor knock-out mice during a new task dissociating strategies of navigation. Journal of Neuroscience, 26(15), 4071-81.
Rupp, J., Blekher, T., Jackson, J., Beristain, X., Marshall, J., Hui, S., ... Foroud, T. (2009). Progression in Prediagnostic Huntington Disease. J Neurol Neurosurg Psychiatry 81(4), 379-384.
Rustay, N. R., Wahlsten, D. & Crabbe, J.C., (2003). Influence of task parameters on rotarod performance and sensitivity to ethanol in mice. Behav Brain Res 141, 237-249.
Salomonczyk, D., Panzera, R., Pirogovosky, E., Goldstein, J., Corey-Bloom, J., Simmons, R. & Gilbert, P. E. (2010). Impaired postural stability as a marker of premanifest Huntington's disease. Mov Disord 25(14), 2428-2433.
Schluter, E. W., Hunsaker, M. R., Greco, C.M., Willemsen, R., Berman, R. F. (2012). Distribution and frequency of intranuclear inclusions in female CGG KI mice modeling the fragile X premutation. Brain Research, 1472, 124-137.
Simon, T. J. (2007). Cognitive characteristics of children with genetic syndromes. Child Adolesc Psychiatr Clin N Am 16, 599-616.
Simon, T. J. (2008). A new account of the neurocognitive foundations of impairments in space, time and number processing in children with chromosome 22q11.2 deletion syndrome. Dev Disabil Res Rev 14, 52-58.
Simon, T. J. (2010). Rewards and challenges of cognitive neuroscience studies of persons with intellectual and developmental disabilities. Am J Intellect Disabilities 115, 79-82.
Simon, T. J. (2011). Clues to the foundation of numerical cognitive impairments: evidence from genetic disorders. Dev Neuropsychol 36(6), 788-805.
Spencer, C. M., Alekseyenko, O., Hamilton, S. M., Thomas, A. M., Serysheva, E., Yuva-Paylor, L. A. & Paylor, R. (2011). Modifying behavioral phenotypes in Fmr1 KO mice: genetic background differences reveal autistic-like responses. Autism Res 4, 40-56.
Tassone, F., Greco, C. M., Hunsaker, M. R., Berman, R. F., Seritan, A. L., Gane, L. W., … Hagerman, R. J. (2011). Neuropathological, clinical, and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes, Brain and Behavior, 11(5), 577-585.
Taylor, T. N., Greene, J. G. & Miller, G. W. (2010). Behavioral phenotyping of mouse models of Parkinson's Disease. Behav Brain Res 211(1), 1-10.
Van Dam, D., Errijgers, V., Kooy, R. F., Willemsen, R., Mientjes, E., Oostra, B. A., & De Deyn, P. P. (2005). Cognitive decline, neuromotor and behavioural disturbances in a mouse model for fragile-X-associated tremor/ataxia syndrome (FXTAS). Behav Brain Res. 162(2), 233-9.
Wahlsten (1974). A developmental time scale for postnatal changes in brain and behavior of B6D2F2 mice. Brain Res 72, 251-264.
Wahlsten, D. (2001). Standardizing tests of mouse behavior: reasons, recommendations & reality. Physiol Behav 73, 695-704.
Wahlsten, D. (1972). Genetic experiments with animal learning: a critical review. Behav Biol 7, 143-182.
Wahlsten, D. (2001). Standardizing tests of mouse behavior: reasons, recommendations & reality. Physiol Behav 73, 695-704.
Wahlsten, D., Bachmanov, A., Finn, D. A. & Crabbe, J. C. (2006). Stability of inbred mouse strain differences in behavior and brain size between laboratories and across decades. Proc Natl Acad Sci 103, 16364-16369.
Wahlsten, D., Metten, Phillips, T. J., Boehm, S. L., Burkhart-Kasch, S., Dorow, J., ... Crabbe, J. C. (2003a). Different data from different labs: lessons from studies of gene-environment interaction. J Neurobiol 54, 283-311.
Wahlsten, D., Rustay, N. R., Metten, P. & Crabbe, J. C. (2003b). In search of a better mouse test. Trends Neurosci 26, 132-136.
Wahlsten, D., Metten, P. & Crabbe, J.C., (2003c). Survey of 21 inbred mouse strains in two laboratories reveals that BTBR T/+ tf/tf has severely reduced hippocampal commissure and absent corpus callosum. Brain Res 971, 47-54.
Wahlsten, D., Bachmanov, A., Finn, D. A. & Crabbe, J. C. (2006). Stability of inbred mouse strain differences in behavior and brain size between laboratories and across decades. Proc Natl Acad Sci, USA 103, 16364-16369.
Wahlsten, D., Metten, P., Phillips, T. J., Boehm, S. L., 2nd, Burkhart-Kasch, S., Dorow, J., ... Crabbe, J. C. (2003). Different data from different labs: lessons from studies of gene-environment interaction. J Neurobiol 54, 283-311.
Wang, Y. C., Magasi, S. R., Bohannon, R. W., Reuben, D. B., McCreath, H. E., Bubela, D. J., ... Rymer, W. Z. (2011). Assessing dexterity function: a comparison of two alternatives for the NIH Toolbox. J Hand Ther. 24(4):313-20.
Wenzel, H. J., Hunsaker, M. R., Greco, C. M., Willemsen, R., & Berman, R. F. (2010). Ubiquitin positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation. Brain Research, 1318, 155-166.
Wesson, D. W., Nixon, R. A., Levy, E. & Wilson, D. A. (2011). Mechanisms of neural and behavioral dysfunction in Alzheimer's disease. Molecular Neurobiology, 43(3), 163-79.
Weiser, M., Van Os, J. & Davidson, M. (2005). Time for a shift in focus in schizophrenia: from narrow phenotypes to broad endophenotypes. B J Psychiatry 187, 203-205.
Whishaw, I. Q. & Tomie, J. (1996). Of mice and mazes: similarities between mice and rats on dry land but not water mazes. Physiology and Behavior, 60(5), 1191-7.
White, N. M. & McDonald, R. J. (2002). Multiple parallel memory systems in the brain of the rat. Neurobiology of Learning and Memory, 77(2), 125-84.
Wilde, E. A., Whiteneck, G. G., Bogner, J., Bushnik, T., Cifu, D. X., Dikmen, S., ... von Steinbuechel, N. (2010). Recommendations for the Use of Common Outcome Measures in Traumatic Brain Injury Research. Arch. Phys. Med. Rehabil. 91(11):1650-1660.
Wong, L. M., Goodrich-Hunsaker, N. J., McLennan, Y. A., Tassone, F., Harvey, D., Rivera, S. M., & Simon, T. J. (2012). Young adult male carriers of the Fragile X premutation exhibit genetically modulated impairments in visuospatial tasks controlled for psychomotor speed. J Neurodev Disord. 4(1), 26.
Xu, B., Karayiorgou, M. & Gogos, J. A. (2010). microRNAs in Psychiatric and Neurodevelopmental Disorders. Brain Res 1338, 78-88.
Yan, Q. J., Asafo-Adjei, P. K., Arnold, H. M., Brown, R. E. & Bauchwitz, R. P. (2004). A phenotypic and molecular characterization of the Fmr1-tm1Cgr fragile X mouse. Genes Brain Behav 3, 337-359.
Yong-Kee, C. J., Salomonczyk, D. & Nash, J. E. (2010). Development and Validation of a Screening Assay for the Evaluation of Putative Neuroprotective Agents in the Treatment of Parkinson's Disease. Neurotox Res 19(4), 519-526.