Robert F. Berman, Jared J. Schwartzer, & Michael R. Hunsaker
Department of Neurological Surgery, Department of Psychiatry and Behavioral Sciences; University of California Davis, Davis, CA, USA
Center for Integrative Neuroscience and Human Behavior; University of Utah; Salt Lake City, UT, USA
ABSTRACT
Fragile X associated tremor/ataxia syndrome (FXTAS) is a late developing neurodegenerative disease that develops in some carriers of the Fragile X premutation (PM). It is the result of an expansion to between 55-200 CGG trinucleotide repeats on the FMR1 gene. CGG KI mouse models have been developed that model much, but not all, of the molecular, histological and neurobehavioral pathology see in FXTAS and in some affected PM carriers. This includes elevated FMR1 mRNA, reduced levels of FMRP, intranuclear inclusions in neurons and astrocytes, mild motor dysfunction resembling ataxia, anxiety and cognitive impairments. These mice have provided insight into when disease processes become evident during development, the molecular mechanisms of pathology, the scope of neurobehavioral involvement, and are being used preclinically to develop and test possible treatment approaches that may improve neurological function in affected individuals. This chapter describes the features of FXTAS, recently recognized pathology in some affected PM carriers, and the development and use of mouse models to study these disorders.
INTRODUCTION
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder characterized by tremors, ataxia, brain atrophy and cognitive decline, and is the result of an CGG trinucleotide repeat expansion in the 5’-untranslated region of the Fragile X mental retardation 1 gene (FMR1). The length of the CGG repeat expansion on FMR1 is polymorphic, with normal repeat lengths ranging between 5-54 (Figure 1). Carriers of a full FMR1 mutation have CGG repeat lengths well over 200, resulting in transcriptional silencing, an absence of the Fragile X mental retardation protein (FMRP), and Fragile X mental retardation syndrome (FXS) (Willemsen et al. 2011). In between are premutation carriers (PM) with CGG repeat lengths between 55-200. Many PM carriers develop neurological and psychological problems over their life spans, including anxiety, depression and poor motor performance (Aziz et al. 2003, Bourgeois et al. 2011, Coffey et al. 2008, Cornish et al. 2005, Farzin et al. 2006, Goodrich-Hunsaker et al. 2011b, Koldewyn et al. 2008, Moore et al. 2004). A subset of older PM carriers is also at risk for developing Fragile X-associated tremor/ataxia syndrome (FXTAS) (Hagerman et al. 2001). Indeed, FXTAS may be one of the more common causes of tremor/ataxia in older adults (Jacquemont et al. 2004a). The prevalence of PM carriers in the general population is estimated to be 1 in 800 males and 1 in 250 females (Hantash et al. 2011, Jacquemont et al. 2004b), so that approximately 1.5 million individuals in the US alone are potentially affected. Because of the dramatic increase in the number of individuals reaching the age of 65, and increasing life expectancy, the numbers of FXTAS patients will increase accordingly, further highlighting the importance of research on PM and FXTAS (Jacquemont et al. 2004a).
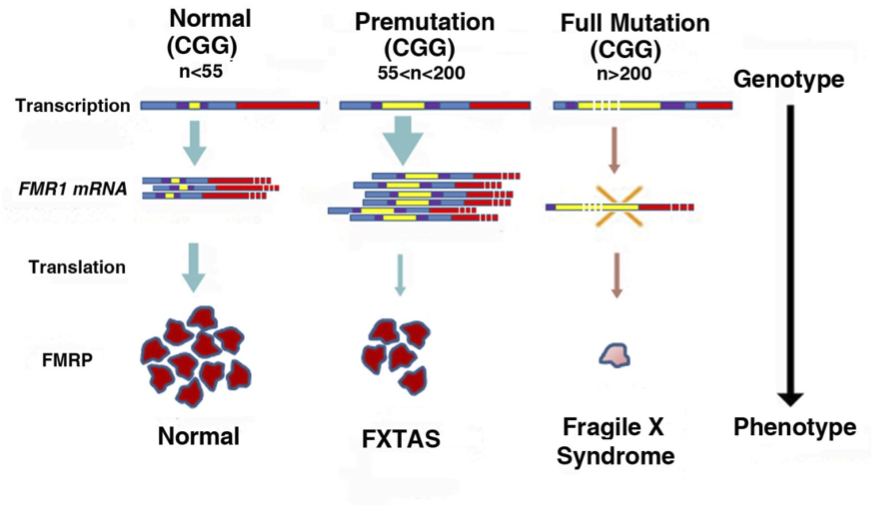 Figure 1: Normal individuals have CGG repeat lengths (yellow segments) on FMR1 between 5-54 CGG repeats. Repeat size in the premutation (PM) range is between 55-200, FMR1 mRNA levels are elevated, FMRP is slightly decreased, and there is increased risk of FXTAS. Repeat size in the full mutation is >200, FMR1 transcription is silenced due to hypermethylation, and the absence of FMRP results in Fragile X syndrome [adapted from Willemsen, et al 2011].
Figure 1: Normal individuals have CGG repeat lengths (yellow segments) on FMR1 between 5-54 CGG repeats. Repeat size in the premutation (PM) range is between 55-200, FMR1 mRNA levels are elevated, FMRP is slightly decreased, and there is increased risk of FXTAS. Repeat size in the full mutation is >200, FMR1 transcription is silenced due to hypermethylation, and the absence of FMRP results in Fragile X syndrome [adapted from Willemsen, et al 2011].
PM carriers were initially thought to be unaffected by their CGG repeat expansion. However, a case study published in 2001 reported that some PM carriers over the age of 50 developed intention tremor, parkinsonism, generalized brain atrophy and showed cognitive decline (Hagerman et al. 2001). Fragile X-associated tremor/ataxia syndrome (FXTAS) was subsequently recognized as a new disease that occurs in a subset of PM carriers. The major symptoms of FXTAS include cerebellar ataxia and intention tremor, often accompanied by lower limb proximal muscle weakness, peripheral neuropathology, memory loss, executive function deficits and autonomic dysfunction, and symmetric white matter lesions of the middle cerebellar peduncle by T2 weighted MRI (i.e., MCP sign) (Jacquemont et al. 2003). Examination of postmortem brains revealed the wide-spread presence of intranuclear inclusions in neurons and astrocytes that stained positive for ubiquitin, as well as Purkinje cell loss and Bergmann gliosis (Greco et al. 2006, Greco et al. 2002). Molecular findings in PM carriers with and without FXTAS include 3-8 fold elevations in FMR1 mRNA in leucocytes and 2-3 fold elevations in brain tissue due to increased FMR1 transcription (Tassone et al. 2007). FMRP levels are reduced by 10-30% due to reduced translational efficiency of FMR1 mRNA bearing a CGG repeat expansion (Primerano et al. 2002). Pathology is currently thought to be due to an RNA toxic “gain of function” (Greco et al. 2002), where elevated levels of mRNA sequester RNA-binding proteins critical for normal cell function, including lamin A/C, Ku80, γH2AX, SAM68, DROSHA and DGCR8 (Sellier et al. 2013, Sellier et al. 2010).
Not all PM carriers develop FXTAS, with penetrance estimated at 17% in male PM carriers between the ages of 50-59 years 38% at 60-69, 47% at 70-79, and up to 75% for carriers over 80 years of age (Jacquemont et al. 2004b). The factors that influence the degree of penetrance remain to be discovered (Hagerman 2012). As a result, a distinction between PM carriers with and without FXTAS has been recognized. The presence of tremor/ataxia is the major diagnostic feature PM carriers who have developed FXTAS, but the majority of PM carriers do not show tremor/ataxia (Aguilar et al. 2008, Narcisa et al. 2011). However, they do show a constellation of other neurobehavioral features, including cognitive deficits, anxiety and depression.
Although originally described in males, symptoms of FXTAS are also seen in some female PM carriers, including intention tremor, cerebellar ataxia, MCP sign, intranuclear inclusions in neurons and astrocytes and hyperintensities in the middle cerebellar peduncle (i.e., MCP sign), although the severity of motor symptoms is generally less than in males as expected in an X-linked disorder (Tassone et al. 2012, Jacquemont et al. 2005). Female PM carriers are also at risk for primary ovarian insufficiency, neuralgia, and thyroid disease (Spath et al. 2011, Hunsaker et al. , Allingham-Hawkins et al. 1999, Cronister et al. 1991). Fragile X-associated primary ovarian insufficiency (FXPOI), results in reduced fertility, irregular menses and early menopause (Vianna-Morgante et al. 1996, Murray 2000, Sherman 2000).
Since those initial reports well over 270 papers have been published describing progressive motor deficits, cognitive impair, neuropathology, and molecular correlates if FXTAS, as briefly reviewed below. The motor deficits initially reported in FXTAS patients included intention tremor, ataxia with wide-based slow gait, muscular rigidity and masked facies (Hagerman et al. 2001). However, many patients later recognized to have FXTAS were originally diagnosed with other movement disorders, including sporadic ataxia, essential tremor (ET) and atypical Parkinsonism (Hall et al. 2006), and this is further complicated by the fact that some FXTAS patients with action tremor and gait ataxia also have symptoms that are consistent with idiopathic Parkinson’s disease (Hall et al. 2009). More recent studies in larger patient populations have confirmed that cerebellar ataxia, action tremor, and parkinsonism are the typical clinical motor features in FXTAS, with an average age of onset in males estimated to be 60.6 (±8.6) years (Leehey et al. 2007). Increasing CGG repeat length and increasing age are strong predictive risk factors for more severe motor features in FXTAS in males and in females, although motor signs are generally milder in women PM carriers (Leehey et al. 2008). Most FXTAS patients develop cerebellar gait ataxia as the disorders progresses, with abnormal tandem gait seen in about 50% of affected males over the age of 50. Signs of Parkinsonism include masked facies, generalized rigidity, slow gait and bradykinesia (Leehey 2009). A detailed analysis of the motor features in older (mean >60 years of age) male PM carriers with and without FXTAS compared to age matched controls was carried out by Aguilar, et al using the CATSYS system (Aguilar et al. 2008). Postural and intention tremor, postural sway, manual coordination and reaction time were analyzed. FXTAS patients showed significantly more postural sway and intention tremor compared to non-FXTAS PM carriers and normal controls, while reaction time, finger tapping and supination-pronation of the hands did not differ significantly among the groups. A similar pattern of findings was reported in females PM carriers with and without FXTAS, although the differences were small and did not reach statistical significance after correction for multiple comparisons (Narcisa et al. 2011). Recent data in 40 FXTAS patients from the CATSYS system to detect mild tremor and ataxia suggested that there may be two phenotypic presentations; a tremor dominant subtype where onset of ataxia is delayed, and an ataxia subtype where ataxia is the dominant feature from the outset (Juncos et al. 2011).
In order to facilitate research on the PM and FXTAS, several animal models have been developed, including drosophila (Jin et al. 2003) and mouse models (Berman and Willemsen 2009, Hunsaker et al. 2012a, Qin et al. 2011), and results from studies in mice are reviewed in this chapter.
CGG KI MOUSE MODEL OF PM AND FXTAS
To investigate the pathological and behavioral consequences of the fragile X PM, a transgenic CGG knock-in (KI) mouse was developed in which the 5’ UTR containing 8 CGG repeats in the native murine FMR1 gene was replaced, via homologous recombination, with a human Nhel–Xhol fragment containing 98 CGG repeats (Willemsen et al. 2003).
| Core Pathology | Human FXTAS | CGG KI Mouse |
|---|---|---|
| CGG Trinucleotide Repeat Length | 55-199 CGG Repeat Length, Repeat Instability | 70-300 CGG Repeats, Modest Repeat Instability |
| Elevated FMR1 mRNA Expression | 2-8 Fold Increases | 1.5-3 Fold Increases |
| FMRP Levels | Reduced in Several Brain Regions | Reduced in Several Brain Regions |
| Motor Impairments | Tremor/Ataxia, Postural Sway, Parkinsonism | Impaired on Rotaros and Ladder Rung Task |
| Cognitive Impairments | Poor Working Memory, Anxiety, Depression< Social Phobia | Spatial Memory Deficits, Anxiety in Plus Maze |
| Intranuclear Inclusions | Neurons and Astrocytes, Highly Correlated with CGG Repeat Length, Frequency Increases with Age | Neurons and Astrocytes, Related to Length of CGG Repeat, Frequency Increases with Age |
Table 1: Comparison of FXTAS with the CGG KI Mouse Model (adapted from Berman & Willemsen, 2009).
Table 1 summarizes the major features associated with FXTAS and the CGG KI mouse model. These KI mice were originally generated on a mixed C57BL/6 X FVB/N background, but CGG KI mice congenic on a C57BL/6 background were subsequently generated and have been used for the majority neurobehavioral studies.
It should be noted that similar to what has been shown in human PM carriers, the CGG KI mouse shows a moderate level of instability across generations in the length of the CGG trinucleotide repeat with a bias toward expansions, particularly upon paternal transmission (Willemsen et al. 2003, Kim 2010). Due to expansion and contraction of the CGG repeat over generations, CGG KI mice with expanded repeat lengths ranging from 70 to >300 have been generated and have provide useful information on how the size of the CGG repeat is related to pathology in the PM and FXTAS. The CGG KI mouse also shows elevated brain levels of FMR1 mRNA as well as moderate reductions in FMRP protein levels compared to wildtype (WT) controls, levels that are further reduced as CGG repeats approach the full mutation range (Brouwer et al. 2008).
Importantly for the development of a mouse model for a genetic disease, the CGG KI mouse provides a useful model for the molecular consequences of the PM, including mitochondrial dysfunction (Kaplan et al. 2012) and alterations to microRNA processing (Sellier et al. 2013). Additionally, the presence of intranuclear inclusions that stain for ubiquitin, the hallmark histopathology in FXTAS, has been shown in neurons, astrocytes and Bergmann glia (Figure 2) in both male and female CGG KI mice (Wenzel et al. 2010, Schluter et al. 2012). The possible contribution of these intranuclear inclusions in the pathophysiology of FXTAS is currently unknown. In addition, intranuclear inclusions are not restricted to the central nervous system, indicating widespread pathology. Intranuclear inclusions are found in many organ systems in in PM carriers, in FXTAS, and also in the CGG KI mouse model, including the pancreas, thyroid, adrenal, pituitary and pineal glands, gastrointestinal system, heart including the mitral value, and are found throughout the autonomic nervous system (Hunsaker et al. 2011). Additional pathological features of FXTAS, including periventricular white matter disease, nodular heterotopia, and white matter hyper-intensities on T2 weighted MRI in the middle cerebellar peduncle (i.e., MCP sign) and pons have not been described in the CGG KI mouse model, and may either not be a feature of this mouse model or may be more difficult to document due to the smaller brain of the mouse making MRI studies difficult and/or the relatively small area of periventricular white matter in mice. In addition, although tremors are an early and cardinal feature of FXTAS, the CGG KI mouse does not appear to develop tremors even when examined at ages greater than 18 months. As indicated above, a distinction between PM carriers with and without FXTAS should be made. The presence of tremor/ataxia is the major diagnostic feature PM carriers who have developed FXTAS, but the majority of carriers do not go on to develop FXTAS and therefore do not show tremor/ataxia (Aguilar et al. 2008, Narcisa et al. 2011). As described above they do show a constellation of other neurobehavioral features, including cognitive deficits, anxiety and depression. Therefore, the mouse CGG KI model appears to model many of the more subtle neurobehavioral features seen in some PM carriers, but lack some important features in FXTAS, with the absence of tremors being most apparent. However, evidence of neuromotor deficits and ataxia associated with poor visuomotor function is found in the CGG KI mice that appear to be relevant to pathology seen in some PM carriers and FXTAS patients, and these are described below.
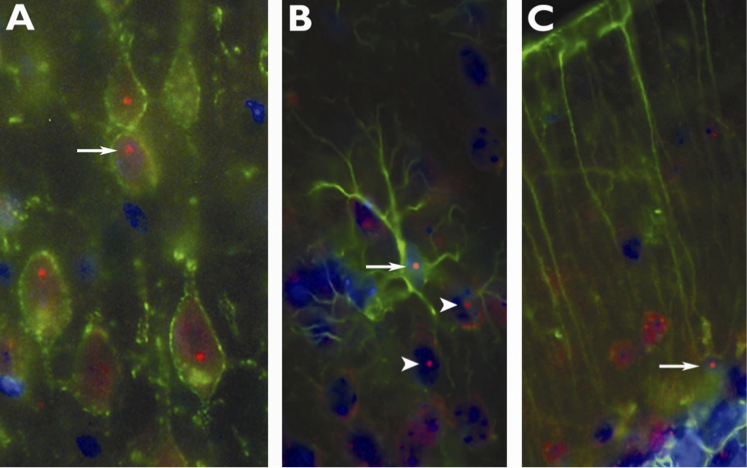 Figure 2: Immunofluorescent photomicrographs of ubiquitin-positive intranuclear inclusions (arrows) immunolabeled red in (A) pyramidal neurons in motor cortex (B) astrocytes and (C) Bergmann glia of CGG KI mice. Neurons (A) were immunolabeled for Kv2.1 potassium channels in the membrane (green), (B) lamina I neocortical astrocytes and (C) Bergmann glia were immunolabeled with GFAP (green). In (B) note several intranuclear inclusions in adjacent neurons (arrowhead). Nuclei were stained with DAPI. (Adapted from Wenzel, et al., 2010).
Figure 2: Immunofluorescent photomicrographs of ubiquitin-positive intranuclear inclusions (arrows) immunolabeled red in (A) pyramidal neurons in motor cortex (B) astrocytes and (C) Bergmann glia of CGG KI mice. Neurons (A) were immunolabeled for Kv2.1 potassium channels in the membrane (green), (B) lamina I neocortical astrocytes and (C) Bergmann glia were immunolabeled with GFAP (green). In (B) note several intranuclear inclusions in adjacent neurons (arrowhead). Nuclei were stained with DAPI. (Adapted from Wenzel, et al., 2010).
Although FXTAS has been considered to be a late onset neurodegenerative disorder, recent findings the CGG KI mouse model provide evidence that pathology may begin much earlier in development than previously thought, as early as embryonic development on E14-17 (Cunningham et al. 2011). Specifically, embryonic CGG KI mice show impaired migration of newborn E17 neurons from the dorsal proliferative zone lining the lateral ventricle to the overlying cortical plate, showing an abnormal polarity compared to WT controls. In addition, the number of Pax6-labeled radial glial cells in the ventricular zone was increased, and the number of Tbr2-labeled intermediate progenitor cells in the subventricular zone was decreased compared to WT embryos, suggesting delayed neuronal differentiation in the embryonic cortex of CGG KI mice. Hippocampal neurons cultured from CGG KI embryos show fewer and shorter dendritic branches and reduced viability in vitro compared to hippocampal neurons from WT mice (Chen 2010). These findings suggest that there may be previously unrecognized brain pathology early in development in FXTAS and in affected PM carriers.
In a further parallel with human disease in PM and FXTAS, female CGG KI mice bearing 130 CGG repeats on FMR1 also show ovarian pathology, including smaller ovarian follicles, fewer granulosa cells, a more rapid loss of follicles during development, compared to WT mice as well as aberrant nuclear accumulation of FMRP in oocytes. Based on these findings in CGG KI mice it has been suggested that FXPOI may be the direct result of pathology within the ovaries, possibly due to accumulation of FMRP (Hoffman et al. 2012).
Another CGG KI mouse model of FXTAS has been developed that shows elevated FMR1 mRNA, intranuclear inclusions and behavioral deficits including hyperactivity, reduced anxiety in the open field and elevated plus maze, poor passive avoidance learning, subtle social interaction deficits but normal motor learning on the rotarod (Qin et al. 2011). These CGG KI mice impaired activity-dependent FMRP translation and enhanced mGluR-dependent long-term depression (LTD) compared to WT mice (Iliff et al. 2013). Therefore, the phenotype of this mouse model appears to overlap with that found in mouse models of FXS. Interestingly, brain levels of FMRP in these mice appear to substantially lower than what has been in reported in PM carriers and in FXTAS patients, as well as lower than levels found in the CGG KI mice developed by the Willemsen laboratory and used in the majority of neurobehavioral studies described in this chapter (Berman and Willemsen 2009, Hunsaker et al. 2012a, Willemsen et al. 2003). It will be important for future studies to determine why these CGG KI mouse models differ in their pathology, including FMRP levels and synaptic plasticity.
CGG KI MOUSE BEHAVIORAL ANALYSES
CGG KI MOUSE VISUOMOTOR FUNCTION
The identification and characterization motor anomalies associated with the fragile X PM have important consequences for understanding the nature of the neuropathology seen in PM carriers, and studies in the CGG KI mouse model have provided important insights in this area. Van dam and colleagues (2005) have identified several motor deficits in CGG KI mice with repeat lengths between 106-123, deficits that became more severe with increased age (Van Dam et al. 2005). Specifically, they reported that 52 and 72 week old male CGG KI mice were unable to stay on an accelerating rotarod as long as younger 20 week old CGG KI mice. In the wire suspension test 52 week old CGG KI mice also fell significantly more often than same age WT mice, although this difference was not significant at 72 weeks of age. CGG KI mice also traveled less in a stationary beam walking test, and showed a reduction in the distance traveled and number of entries into the center of an open field apparatus. Although these data suggest neuromotor deficits in CGG KI mice, more sensitive measures of motor function that more directly model the behavioral measure in humans (i.e., CATSYS) are required.
To specifically evaluate visuomotor functioning CGG KI mice, a skilled forelimb reaching task was developed by modifying existing protocols to emphasize the acquisition of skilled reaching performance rather than performance per se (Diep et al. 2012). In this task, CGG KI and WT mice were required to reach through a narrow slit-window in the front of a transparent Plexiglas box to obtain a food reward pellet (i.e., 45 mg sucrose pellet, Bioserve, Beltsville, MD) positioned just out of reach and at a 30 degree angle off the edge of the slit-window. Mice were required to reach with their non-preferred paw. The number of pellets the mouse obtained along with errors, including dropping the pellet or knocking it away, were recorded. The CGG KI mice showed a slower learning curve than wild type mice, with CGG KI mice learning the task on average 1 day later than WT litter mates and never reaching the same level of performance as WT mice (Diep et al. 2012). Importantly, these forelimb motor learning deficits were subtle, only becoming apparent when the mice were required to perform a difficult reaching task. There were no observed differences in these mice in measures of grip strength measures or other measures of limb usage. In preliminary studies the qualitative data suggests that the CGG KI mice reached with more of a circular or radial motion rather than a directed vector toward the reward pellet. This appears to result in the CGG KI mice knocking the reward away or having difficulty in grasping the reward (Figure 3). Once the CGG KI mice grasped the reward pellet, however, they were typically able to consume it, not showing any difference in the ability to hold onto the pellet and consume it.
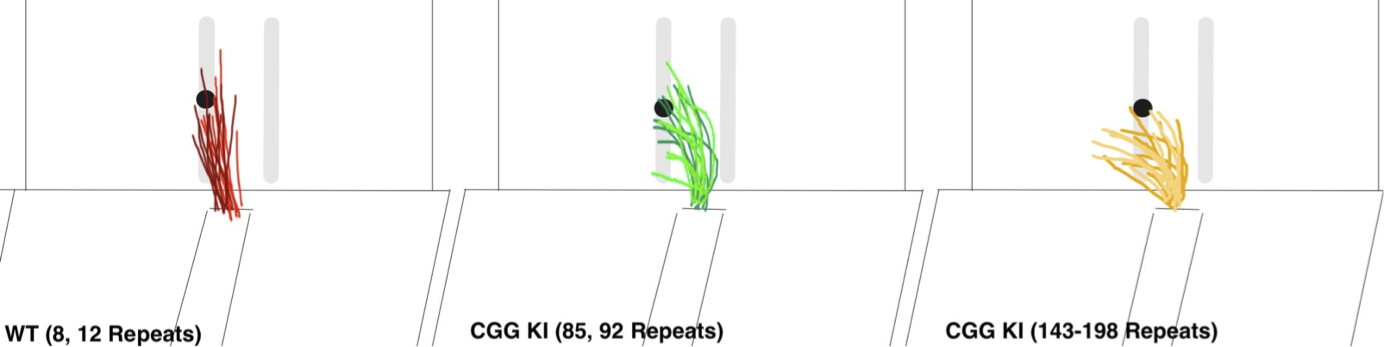 Figure 3: Traces for reaching trajectories over 10 consecutive reaches for 2 wildtype mice (WT), 2 low CGG KI mice (CGG repeats = 85, 92) and 2 high CGG KI mice (CGG repeats = 143, 198). Note the increasingly radial trajectories in mice with expanded CGG repeats compared to WT mice.
Figure 3: Traces for reaching trajectories over 10 consecutive reaches for 2 wildtype mice (WT), 2 low CGG KI mice (CGG repeats = 85, 92) and 2 high CGG KI mice (CGG repeats = 143, 198). Note the increasingly radial trajectories in mice with expanded CGG repeats compared to WT mice.
Damaged to the forelimb motor area impairs performance in a similar forelimb skilled reaching task in mice (Zeiler et al. 2013), and training in a forelimb reaching task results in rapid formation and stabilization of synapses in motor cortex (Xu et al. 2009). Because CGG KI mice show abnormal cortical dendrite and dendritic spine morphology (Berman et al. 2012, Qin et al. 2011) (Figure 4), it is possible that poor learning and performance in the reaching task in CGG KI mice is a result of pathology in motor cortex . Another possibility is that the pontocerebellar motor system is abnormal. This is supported by histological and MRI data indicating the presence of white matter disease in the middle cerebellar peduncle and adjacent cerebellar white matter in FXTAS patients ((Greco et al. 2006, Brunberg et al. 2002). The cerebellum plays a critical role in learning sensorimotor tasks, and the pontocerebellar tract is a key pathway contributing to new motor skills (Manto and Jissendi 2012, Ito 2006), so that cerebellar pathology could underlie the impairment in fine motor skills required to skillfully reach, grasp, and consume the food rewards. It is also possible that the parietal cortex through its interactions with the superior colliculus and cerebellum is unable to provide adequate spatiotemporal updating to allow the CGG KI mice to reach the same level of performance as WT mice. This interpretation is supported by recent experiments demonstrating superior colliculus activation during reaching in humans (Linzenbold et al. 2011).
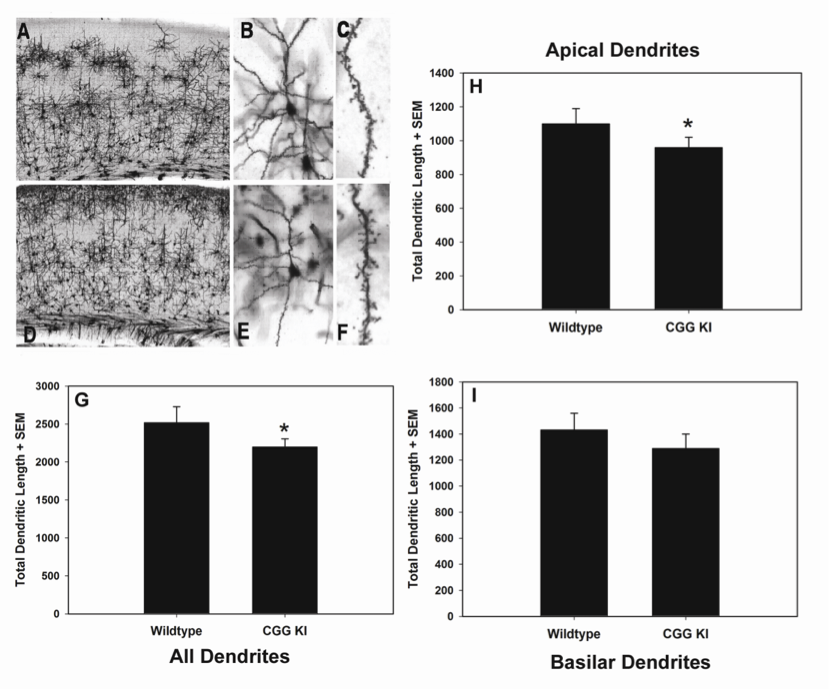 Figure 4: Representative photomicrographs of Golgi-Cox stained pyramidal neurons in layer II/III of primary visual cortex of WT (A, B, C) and CGG KI mice (D, E, F) at progressively higher magnification. Total dendritic length was calculated from reconstructed dendritic arbors using Neurolucida software. Total dendritic length (G) was reduced for all dendrites (p < 0.05), but was only significantly reduced for apical dendrites (p < 0.05) when apical (H) and basilar (I) dendrites were analyzed separately. Magnification: A &D– 4x, B & E – 20x, C & F – 100x. Reprinted with permission Epilepsia ILAE
Figure 4: Representative photomicrographs of Golgi-Cox stained pyramidal neurons in layer II/III of primary visual cortex of WT (A, B, C) and CGG KI mice (D, E, F) at progressively higher magnification. Total dendritic length was calculated from reconstructed dendritic arbors using Neurolucida software. Total dendritic length (G) was reduced for all dendrites (p < 0.05), but was only significantly reduced for apical dendrites (p < 0.05) when apical (H) and basilar (I) dendrites were analyzed separately. Magnification: A &D– 4x, B & E – 20x, C & F – 100x. Reprinted with permission Epilepsia ILAE
To evaluate potential subclinical gait ataxia or general clumsiness in the CGG KI mice, a skilled ladder-walking task has been employed. In this test mice are allowed to walk across a series of very thin ladder-rungs perpendicular to the direction of travel (similar to walking across a ladder set on the ground). The number of times that the mouse makes an error in foot placement was recorded and was operationally defined as a foot slip. To perform these tasks, the mouse was placed at one end of the apparatus and allowed to cross from one end to the other into a darkened box.
The apparatus used in these experiments was modified from previously described ladder rung task (Gonzalez et al. 2004). The apparatus had black plastic walls separated by approximately 5 cm, with 2 mm diameter rungs making up the floor. For this initial study, the mice were placed at one end of the apparatus and were allowed to walk back and forth for 2 minutes. The number of foot slips were recorded for the 2 min duration, except for when the mouse was turning around. The number of times the mouse traveled from one end of the apparatus to the other was also recorded as a general locomotor measure. On this task, CGG KI mice as young as 2 months of age already showed an increased number of foot slips compared to WT litter mate controls. Importantly, the CGG KI mice showed more of both forelimb and hindlimb slips than WT mice, suggesting both visuospatial and basic motor deficits. That is, forelimb foot slips may suggest visuospatial processing deficits, such as difficulty in planning forepaw placement as well as difficulty updating movements as the step progressed (i.e., as the mouse moved forward the initial planned step has to be modified subtly and an inability to do so results in a foot slip). Hind foot slips however, have less of a visuospatial planning component, but may reflect a subtle motor effect. This is because stepping with the hind limbs has been shown to be more efficient and easier for the mouse to perform (Metz and Whishaw 2002). Difficulty in hind limb stepping may therefore reflect some form of ataxia, and may underlie impairments in rotarod and stationary beam walking reported previously (Van Dam et al. 2005).
A group at Erasmus MC in Rotterdam developed a skilled walking task called the Erasmus ladder, where the testing and collection of data are fully automated (van der Vaart et al. 2011). Similar to the ladder rung apparatus described above, the Erasmus ladder has a series of horizontal ladder-steps that the mouse must traverse in order to enter an escape/start box at the end. The apparatus was designed to use puffs of air to induce the mouse to shuttle between the two boxes by crossing the ladder steps. The system automatically captures step times, foot slips, misplacement of limbs, etc. Cupido (2009) found that CGG KI mice showed a significantly increased number of foot slips and missteps (Figure 5), although they did not differ from WT mice in step time, suggesting intact general motor function (Cupido 2009). Additionally, the CGG KI mice were able to perform the motor associative learning task as well as wild type litter mate control animals. In contrast, FMR1 KO mice showed a deficit for the associative learning task, never performing as well as CGG KI and WT control animals, while the number of footslips and missteps did not differ from WT controls, suggesting intact visuospatial, visuomotor, and basic motor functions.
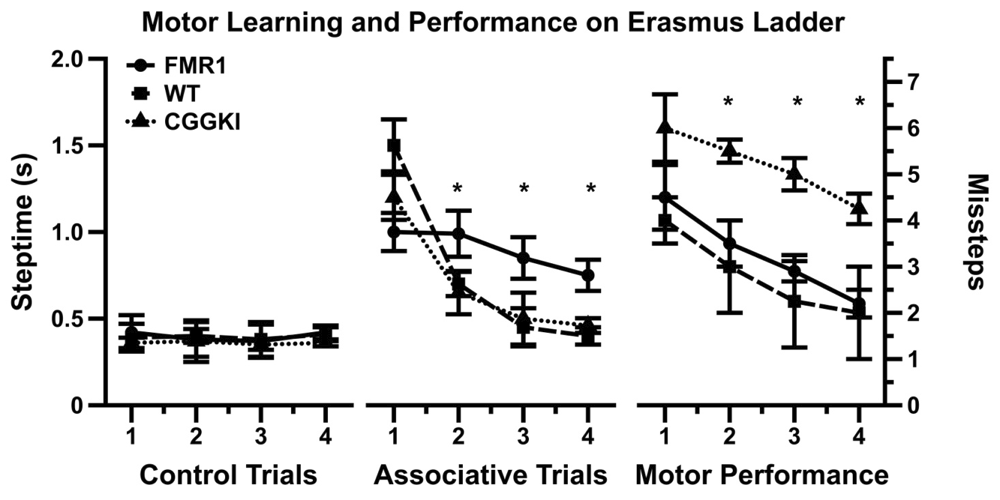 Figure 5: Data redrawn from Cupido (2009) demonstrating the dissociation between motor performance and motor associative learning in CGG KI mice and the Fmr1 KO mouse models of fragile x syndrome.
Figure 5: Data redrawn from Cupido (2009) demonstrating the dissociation between motor performance and motor associative learning in CGG KI mice and the Fmr1 KO mouse models of fragile x syndrome.
It has been demonstrated that visuomotor function depends on spatiotemporal integration (Sailer et al. 2000). This means that for an individual to act in space, he must be able to (1) select a goal of action and (2) plan the movements, (3) initiate the movements, (4) modify the ongoing movement using updated spatiotemporal information, and (5) properly terminate the movement as intended. Intact spatiotemporal updating prevents misguided movements, such as reaching for a cup and knocking it off the table because an inability to stop or slow the progress of the hand. This is increasingly important if reaching or acting in space extends to processing angles among stimuli or targets that change over time. This updating process is what may be deficient in PM carriers and in FXTAS patients, and in the CGG KI mice as they show similar spatiotemporal processing deficits and thus reduced spatial and temporal resolution (Kraan et al. 2013). This type of spatiotemporal deficits may underlie anecdotal reports of subclinical apraxia and general clumsiness among premutation carriers.
CGG KI MOUSE SPATIOTEMPORAL PROCESSING
The CGG KI mouse also shows deficits in processing of spatial and temporal information that is also associated with impaired hippocampal synaptic plasticity, including reduced LTP and LTD (Hunsaker et al. 2012b). This includes spatial memory deficits on the Morris water maze in KI mice older than 52 weeks of age (Van Dam et al. 2005). These deficits, however, appear to be very mild and not nearly as profound as the general memory deficits demonstrated in the most severely affected FXTAS patients (Hagerman et al. 2009). More recent studies have examined spatial and temporal processing deficits in CGG KI mice using behavioral tasks designed to test specific hypotheses about the cognitive strengths and weaknesses reported in fragile X PM carriers without FXTAS (Goodrich-Hunsaker et al. 2011a, Goodrich-Hunsaker et al. 2011b, Goodrich-Hunsaker et al. 2011c, Wong et al. 2012). These cognitive domains were chosen because a number of neurodevelopmental disorders share fundamental spatiotemporal processing deficits described as a form of spatiotemporal “hypergranularity” (Simon 2007, Simon 2008, Simon 2011). This hypergranularity results in impaired spatiotemporal memory resolution, such that a greater difference among elements in spatial or temporal separation is required before they can be discriminated. (Hunsaker 2012b, Hunsaker 2012a, Simon 2007, Simon 2008, Simon 2011)
Using a pair of behavioral tasks to evaluate spatial resolution of spatial memory in CGG KI mice, it was demonstrated that CGG KI mice show deficits for processing the specific distances that separate two objects in space using a “metric” change detection task (also called a coordinate change). These deficits were present as early as 3 months of age, and persisted until at least 12 months of age (Hunsaker et al. 2012b, Hunsaker et al. 2009). Performance also tested in a "topological” spatial memory task that required the mice to remember the relative spatial locations of two dissimilar objects after their positions were transposed (called a topological or categorical change). CGG KI mice did not show deficits at 3 or 6 months of age, but did show deficits when they were 9 and 12 months of age compared to age matched WT littermate controls (Hunsaker et al. 2012b, Hunsaker et al. 2009). Findings from these two studies suggest that: (1) that the resolution of spatial processing in CGG KI mice is reduced from a very young age, and this reduced spatial resolution appears to be then be maintained over time, such that the resolution does not progressively worsen with aging; (2) general spatial memory as measured by the topological change detection task does show a progressive worsening across age, with deficits emerging in adults and worsening with advancing age in CGG KI mice, a pattern similar to the deficits seen in the water maze (Hunsaker et al. 2012b, Hunsaker et al. 2009, Van Dam et al. 2005).
To evaluate temporal memory in CGG KI mice, a temporal ordering for visual objects task was used (Hunsaker et al. 2010). In this task, mice are presented sequentially with three different pairs of identical objects, and time spent exploring the objects is recorded. They are then presented with one object from the first pair and one object from the third pair, and time spent exploring is again recorded. Typically, mice preferentially explore the first over the third object. On another day after a different set of objects have been presented, the mice are presented with the first object they were presented that day as well as a never before seen novel object. Mice preferentially explore the novel object over the familiar one. On these tests the CGG KI mice show more exploration of the novel object as do WT’s, but unlike WT’s do not show an object preference in the temporal ordering test (i.e., tested with first and third presented object). This pattern of results indicates that object recognition is intact, but the processing of temporal order is impaired.
An important aspect of these behavioral results is that both male and female CGG KI mice showed deficits. This is important because female premutation carriers generally show reduced disease severity due to the protective effect of a second, non-mutated FMR1 gene on the inactive X allele (Berry-Kravis et al. 2005, Berry-Kravis et al. 2007, Schluter et al. 2012, Tassone et al. 2012). The presence of these deficits in both male and female CG KI mice suggests impairments to these processes are fundamental consequences of the premutation, since deficits are present and identifiable even in the least affected of the population similar effects are currently being identified and characterized in human premutation carriers both symptomatic and asymptomatic for FXTAS (Aguilar et al. 2008, Hagerman et al. 2001, Narcisa et al. 2011, Allen et al. 2008).
COMPARING HUMAN PATHOLOGY TO DEFICITS IN MOUSE MODELS
It is important for mouse models to be relevant to the clinical manifestations of disease they are designed to model. However, no mouse model can completely capture all of the features of a clinical disorder, including the fragile X PM and FXTAS. In addition, there are obvious differences in the research protocols used in animal models and clinical research. However, there are similarities between the behavioral characteristics of the CGG KI mouse and features seen in PM carriers and in FXTAS that make the CGG KI mouse useful. For example, in some FXTAS patients the ataxia becomes more pronounced as the affected individual walks until they lose balance (Berry-Kravis et al. 2007). They appear relatively normal for the first few steps, but then a postural sway emerges that grows in amplitude with each step until the patient either braces against a wall or falls over. For the intention tremor, the FXTAS patients appear to show normal motor function at first, but as the trial continues (i.e., spiral of Archimedes or drawing a line between two lines), small oscillations emerge which increases amplitude until the patient stops performing (Peters et al. 2006). These impaired motor behaviors suggest abnormal feedback among cortical and cerebellar systems that impair online corrections of movements so that errors accumulate and increase in severity as movement progresses.
Although tremors are not an obvious feature of the CGG KI mouse model, a possible analogy to the oscillatory motor behaviors in FXTAS may be found in the skilled reaching task. When the CGG KI mice were reaching for the reward pellets, they often reached with a slightly circuitous path, not a direct vector to the pellet. If they missed the pellet, the subsequent reaches took an increasingly wide and uncontrolled pattern that continued across 3-5 reaches until the mouse knocked the pellet off the platform (cf., Figure 3). When the wild type mice missed the pellet, they continued reaching using a direct vector to the pellet until they retrieved it, which they usually were able to. Only limited preliminary data are available to support this conjecture (data from 2 CGG KI and 2 WT litter mates are provided in figure 3 as vector sketches recorded off video recorded trials). Therefore, future work will be needed to develop methods to video-capture reaching trials at much higher temporal resolution (Gonzalez et al. 2004, Whishaw et al. 1991).
FUTURE DIRECTIONS
Despite the ability to identify and characterize visuomotor deficits in CGG KI mice, the widespread neuropathology (e.g., intranuclear inclusions, dendrite and spine dysmorphology) makes it difficult to determine whether these motor deficits derive from dysfunction in cerebral, cerebellar or other circuits. Both the skilled reaching data as well as the skilled walking deficits could also be explained by cortical (i.e., motor cortex, parietal cortex) dysfunction, or dysfunction in pontocerebellar or cerebellar circuits. A likely possibility based on data from the CGG KI mouse, is that the distributed cortico-cerebellar circuits described by Diep et al. (2012) are altered and may underlie these deficits (Diep et al. 2012). However, this distributed network explanation actually raises more issues than it answers. For one, do disruptions in one node of the network (i.e., the inferior parietal lobe) result in qualitatively and quantitatively similar deficits as cerebellar or collicular disruptions? To date these questions remain unanswered.
One way to evaluate the differential contributions of these different networks is to apply transgenic mouse technologies to the problem. Hashem et al. (2009) demonstrated that a premutation length CGG repeat expressed specifically in Purkinje cell populations using an L7/pcp2 promoter resulted in Purkinje cell loss and major impairments on the accelerated rotarod (Hashem et al. 2009). These data were interpreted as ataxia like gait disturbances, although no explicit analyses of fine motor function were attempted. An analysis of these mice on skilled reaching performance as well as skilled ladder rung walking or on the Erasmus ladder in future studies may help to reveal the importance of cerebellar pathology for the gait distances associated with the PM and seen in FXTAS. Similarly, development of inducible CGG KI mice, where the expression of a CGG repeat expansion is under experimenter control (e.g., dox-inducible) is underway in our laboratory. By using cell-specific promoters we are selectively expressing a CGG98 repeat in astrocytes, neurons of the forebrain or in cerebellar Purkinje cell using Gfa2, CamKIIα and L7/pcp2 promoters, respectively. The hope is that by comparing pathology in these types of models we will better understand the underlying cellular disease mechanisms in PM and FXTAS. Such data would be critical to inform and target potential treatments to relevant brain regions and cell classes.
Conclusions
FXTAS is a recently described disorder due to a CGG repeat expansion on FMR1. Although originally recognized based on the presence of tremor and gait ataxia in older relatives of individuals with Fragile X syndrome, the recognized features in FXTAS have now expanded to include hallmark brain pathology (e.g., intranuclear inclusions, white matter disease), molecular findings (e.g., elevated FMR1 mRNA, decreased levels of FMRP), and neurocognitive impairments. In addition, many studies have now identified subtle motor and cognitive effects in otherwise asymptomatic PM carriers. As a result, the disorder should be viewed as more complex than implied by the “tremor/ataxia” in the syndrome’s name. Development of mouse models, and specifically the CGG KI mouse, has allowed for the study of the PM and FXTAS at many levels, including anatomical, molecular and behavioral. The existing mouse models are useful, but in need of further development in order to better model some key features of FXTAS including intentional tremors. Development of new transgenic models should allow more specific questions to be asked about these disorders, including the necessity and sufficiency of specific cell involvement for pathology, as well as a brain regional analysis of the circuitry that may underlie pathology.
References
Aguilar, D., Sigford, K. E., Soontarapornchai, K., Nguyen, D. V., Adams, P. E., Yuhas, J. M., Tassone, F., Hagerman, P. J. and Hagerman, R. J. (2008) 'A quantitative assessment of tremor and ataxia in FMR1 premutation carriers using CATSYS', Am J Med Genet A, 146A(5), 629-35.
Allen, E. G., Juncos, J., Letz, R., Rusin, M., Hamilton, D., Novak, G., Shubeck, L., Tinker, S. W. and Sherman, S. L. (2008) 'Detection of early FXTAS motor symptoms using the CATSYS computerised neuromotor test battery', J Med Genet, 45(5), 290-7.
Allingham-Hawkins, D. J., Babul-Hirji, R., Chitayat, D., Holden, J. J., Yang, K. T., Lee, C., Hudson, R., Gorwill, H., Nolin, S. L., Glicksman, A., Jenkins, E. C., Brown, W. T., Howard-Peebles, P. N., Becchi, C., Cummings, E., Fallon, L., Seitz, S., Black, S. H., Vianna-Morgante, A. M., Costa, S. S., Otto, P. A., Mingroni-Netto, R. C., Murray, A., Webb, J., Vieri, F. and et al. (1999) 'Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study--preliminary data', Am J Med Genet, 83(4), 322-5.
Aziz, M., Stathopulu, E., Callias, M., Taylor, C., Turk, J., Oostra, B., Willemsen, R. and Patton, M. (2003) 'Clinical features of boys with fragile X premutations and intermediate alleles', Am J Med Genet B Neuropsychiatr Genet, 121(1), 119-27.
Berman, R. F., Murray, K. D., Arque, G., Hunsaker, M. R. and Wenzel, H. J. (2012) 'Abnormal dendrite and spine morphology in primary visual cortex in the CGG knock-in mouse model of the fragile X premutation', Epilepsia, 53 Suppl 1, 150-60.
Berman, R. F. and Willemsen, R. (2009) 'Mouse models of fragile x-associated tremor ataxia', J Investig Med, 57(8), 837-41.
Berry-Kravis, E., Abrams, L., Coffey, S. M., Hall, D. A., Greco, C., Gane, L. W., Grigsby, J., Bourgeois, J. A., Finucane, B., Jacquemont, S., Brunberg, J. A., Zhang, L., Lin, J., Tassone, F., Hagerman, P. J., Hagerman, R. J. and Leehey, M. A. (2007) 'Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines', Mov Disord, 22(14), 2018-30, quiz 2140.
Berry-Kravis, E., Potanos, K., Weinberg, D., Zhou, L. and Goetz, C. G. (2005) 'Fragile X-associated tremor/ataxia syndrome in sisters related to X-inactivation', Ann Neurol, 57(1), 144-7.
Bourgeois, J. A., Seritan, A. L., Casillas, E. M., Hessl, D., Schneider, A., Yang, Y., Kaur, I., Cogswell, J. B., Nguyen, D. V. and Hagerman, R. J. (2011) 'Lifetime prevalence of mood and anxiety disorders in fragile X premutation carriers', J Clin Psychiatry, 72(2), 175-82.
Brouwer, J. R., Huizer, K., Severijnen, L. A., Hukema, R. K., Berman, R. F., Oostra, B. A. and Willemsen, R. (2008) 'CGG-repeat length and neuropathological and molecular correlates in a mouse model for fragile X-associated tremor/ataxia syndrome', J Neurochem, 107(6), 1671-82.
Brunberg, J. A., Jacquemont, S., Hagerman, R. J., Berry-Kravis, E. M., Grigsby, J., Leehey, M. A., Tassone, F., Brown, W. T., Greco, C. M. and Hagerman, P. J. (2002) 'Fragile X premutation carriers: characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction', AJNR Am J Neuroradiol, 23(10), 1757-66.
Chen, Y., Tassone, F., Berman, R.F., Hagerman, P.M., Hagerman, R.J., Willemsen, R. & Pessah, I.N. (2010) 'Murine hippocampal neurons expressing FMR1 gene premutation show early developmental deficits and late degeneration.', Human Molecular Genetics, 19(1), 196-208.
Coffey, S. M., Cook, K., Tartaglia, N., Tassone, F., Nguyen, D. V., Pan, R., Bronsky, H. E., Yuhas, J., Borodyanskaya, M., Grigsby, J., Doerflinger, M., Hagerman, P. J. and Hagerman, R. J. (2008) 'Expanded clinical phenotype of women with the FMR1 premutation', Am J Med Genet A, 146A(8), 1009-16.
Cornish, K., Kogan, C., Turk, J., Manly, T., James, N., Mills, A. and Dalton, A. (2005) 'The emerging fragile X premutation phenotype: evidence from the domain of social cognition', Brain Cogn, 57(1), 53-60.
Cronister, A., Schreiner, R., Wittenberger, M., Amiri, K., Harris, K. and Hagerman, R. J. (1991) 'Heterozygous fragile X female: historical, physical, cognitive, and cytogenetic features', Am J Med Genet, 38(2-3), 269-74.
Cunningham, C. L., Martinez Cerdeno, V., Navarro Porras, E., Prakash, A. N., Angelastro, J. M., Willemsen, R., Hagerman, P. J., Pessah, I. N., Berman, R. F. and Noctor, S. C. (2011) 'Premutation CGG-repeat expansion of the FMR1 gene impairs mouse neocortical development', Hum Mol Genet, 20(1), 64-79.
Cupido, A. (2009) 'Detecting cerebellar phenotypes with the Erasmus ladder. Ph.D. Thesis, Erasmus University, Rotterdam, Netherlands.'.
Diep, A. A., Hunsaker, M. R., Kwock, R., Kim, K., Willemsen, R. and Berman, R. F. (2012) 'Female CGG knock-in mice modeling the fragile X premutation are impaired on a skilled forelimb reaching task', Neurobiol Learn Mem, 97(2), 229-34.
Farzin, F., Perry, H., Hessl, D., Loesch, D., Cohen, J., Bacalman, S., Gane, L., Tassone, F., Hagerman, P. and Hagerman, R. (2006) 'Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation', J Dev Behav Pediatr, 27(2 Suppl), S137-44.
Gonzalez, C. L., Gharbawie, O. A., Williams, P. T., Kleim, J. A., Kolb, B. and Whishaw, I. Q. (2004) 'Evidence for bilateral control of skilled movements: ipsilateral skilled forelimb reaching deficits and functional recovery in rats follow motor cortex and lateral frontal cortex lesions', Eur J Neurosci, 20(12), 3442-52.
Goodrich-Hunsaker, N. J., Wong, L. M., McLennan, Y., Srivastava, S., Tassone, F., Harvey, D., Rivera, S. M. and Simon, T. J. (2011a) 'Young adult female fragile X premutation carriers show age- and genetically-modulated cognitive impairments', Brain Cogn, 75(3), 255-60.
Goodrich-Hunsaker, N. J., Wong, L. M., McLennan, Y., Tassone, F., Harvey, D., Rivera, S. M. and Simon, T. J. (2011b) 'Adult Female Fragile X Premutation Carriers Exhibit Age- and CGG Repeat Length-Related Impairments on an Attentionally Based Enumeration Task', Front Hum Neurosci, 5, 63.
Goodrich-Hunsaker, N. J., Wong, L. M., McLennan, Y., Tassone, F., Harvey, D., Rivera, S. M. and Simon, T. J. (2011c) 'Enhanced manual and oral motor reaction time in young adult female fragile X premutation carriers', J Int Neuropsychol Soc, 17(4), 746-50.
Greco, C. M., Berman, R. F., Martin, R. M., Tassone, F., Schwartz, P. H., Chang, A., Trapp, B. D., Iwahashi, C., Brunberg, J., Grigsby, J., Hessl, D., Becker, E. J., Papazian, J., Leehey, M. A., Hagerman, R. J. and Hagerman, P. J. (2006) 'Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS)', Brain, 129(Pt 1), 243-55.
Greco, C. M., Hagerman, R. J., Tassone, F., Chudley, A. E., Del Bigio, M. R., Jacquemont, S., Leehey, M. and Hagerman, P. J. (2002) 'Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers', Brain, 125(Pt 8), 1760-71.
Hagerman, P. J. (2012) 'Current Gaps in Understanding the Molecular Basis of FXTAS', Tremor Other Hyperkinet Mov (N Y), 2.
Hagerman, R. J., Berry-Kravis, E., Kaufmann, W. E., Ono, M. Y., Tartaglia, N., Lachiewicz, A., Kronk, R., Delahunty, C., Hessl, D., Visootsak, J., Picker, J., Gane, L. and Tranfaglia, M. (2009) 'Advances in the treatment of fragile X syndrome', Pediatrics, 123(1), 378-90.
Hagerman, R. J., Leehey, M., Heinrichs, W., Tassone, F., Wilson, R., Hills, J., Grigsby, J., Gage, B. and Hagerman, P. J. (2001) 'Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X', Neurology, 57(1), 127-30.
Hall, D. A., Hagerman, R. J., Hagerman, P. J., Jacquemont, S. and Leehey, M. A. (2006) 'Prevalence of FMR1 repeat expansions in movement disorders. A systematic review', Neuroepidemiology, 26(3), 151-5.
Hall, D. A., Howard, K., Hagerman, R. and Leehey, M. A. (2009) 'Parkinsonism in FMR1 premutation carriers may be indistinguishable from Parkinson disease', Parkinsonism Relat Disord, 15(2), 156-9.
Hantash, F. M., Goos, D. M., Crossley, B., Anderson, B., Zhang, K., Sun, W. and Strom, C. M. (2011) 'FMR1 premutation carrier frequency in patients undergoing routine population-based carrier screening: insights into the prevalence of fragile X syndrome, fragile X-associated tremor/ataxia syndrome, and fragile X-associated primary ovarian insufficiency in the United States', Genet Med, 13(1), 39-45.
Hashem, V., Galloway, J. N., Mori, M., Willemsen, R., Oostra, B. A., Paylor, R. and Nelson, D. L. (2009) 'Ectopic expression of CGG containing mRNA is neurotoxic in mammals', Hum Mol Genet, 18(13), 2443-51.
Hoffman, G. E., Le, W. W., Entezam, A., Otsuka, N., Tong, Z. B., Nelson, L., Flaws, J. A., McDonald, J. H., Jafar, S. and Usdin, K. (2012) 'Ovarian abnormalities in a mouse model of fragile X primary ovarian insufficiency', J Histochem Cytochem, 60(6), 439-56.
Hunsaker, M. R. (2012a) 'Comprehensive neurocognitive endophenotyping strategies for mouse models of genetic disorders', Prog Neurobiol, 96(2), 220-41.
Hunsaker, M. R. (2012b) 'The importance of considering all attributes of memory in behavioral endophenotyping of mouse models of genetic disease', Behav Neurosci, 126(3), 371-80.
Hunsaker, M. R., Arque, G., Berman, R. F., Willemsen, R. and Hukema, R. K. (2012a) 'Mouse models of the fragile x premutation and the fragile X associated tremor/ataxia syndrome', Results Probl Cell Differ, 54, 255-69.
Hunsaker, M. R., Goodrich-Hunsaker, N. J., Willemsen, R. and Berman, R. F. (2010) 'Temporal ordering deficits in female CGG KI mice heterozygous for the fragile X premutation', Behav Brain Res, 213(2), 263-8.
Hunsaker, M. R., Greco, C. M., Spath, M. A., Smits, A. P., Navarro, C. S., Tassone, F., Kros, J. M., Severijnen, L. A., Berry-Kravis, E. M., Berman, R. F., Hagerman, P. J., Willemsen, R., Hagerman, R. J. and Hukema, R. K. 'Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice', Acta Neuropathol.
Hunsaker, M. R., Greco, C. M., Spath, M. A., Smits, A. P., Navarro, C. S., Tassone, F., Kros, J. M., Severijnen, L. A., Berry-Kravis, E. M., Berman, R. F., Hagerman, P. J., Willemsen, R., Hagerman, R. J. and Hukema, R. K. (2011) 'Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice', Acta Neuropathol, 122(4), 467-79.
Hunsaker, M. R., Kim, K., Willemsen, R. and Berman, R. F. (2012b) 'CGG trinucleotide repeat length modulates neural plasticity and spatiotemporal processing in a mouse model of the fragile X premutation', Hippocampus.
Hunsaker, M. R., Wenzel, H. J., Willemsen, R. and Berman, R. F. (2009) 'Progressive spatial processing deficits in a mouse model of the fragile X premutation', Behav Neurosci, 123(6), 1315-24.
Iliff, A. J., Renoux, A. J., Krans, A., Usdin, K., Sutton, M. A. and Todd, P. K. (2013) 'Impaired activity-dependent FMRP translation and enhanced mGluR-dependent LTD in Fragile X premutation mice', Hum Mol Genet, 22(6), 1180-92.
Ito, M. (2006) 'Cerebellar circuitry as a neuronal machine', Prog Neurobiol, 78(3-5), 272-303.
Jacquemont, S., Farzin, F., Hall, D., Leehey, M., Tassone, F., Gane, L., Zhang, L., Grigsby, J., Jardini, T., Lewin, F., Berry-Kravis, E., Hagerman, P. J. and Hagerman, R. J. (2004a) 'Aging in individuals with the FMR1 mutation', Am J Ment Retard, 109(2), 154-64.
Jacquemont, S., Hagerman, R. J., Leehey, M., Grigsby, J., Zhang, L., Brunberg, J. A., Greco, C., Des Portes, V., Jardini, T., Levine, R., Berry-Kravis, E., Brown, W. T., Schaeffer, S., Kissel, J., Tassone, F. and Hagerman, P. J. (2003) 'Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates', Am J Hum Genet, 72(4), 869-78.
Jacquemont, S., Hagerman, R. J., Leehey, M. A., Hall, D. A., Levine, R. A., Brunberg, J. A., Zhang, L., Jardini, T., Gane, L. W., Harris, S. W., Herman, K., Grigsby, J., Greco, C. M., Berry-Kravis, E., Tassone, F. and Hagerman, P. J. (2004b) 'Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population', Jama, 291(4), 460-9.
Jacquemont, S., Orrico, A., Galli, L., Sahota, P. K., Brunberg, J. A., Anichini, C., Leehey, M., Schaeffer, S., Hagerman, R. J., Hagerman, P. J. and Tassone, F. (2005) 'Spastic paraparesis, cerebellar ataxia, and intention tremor: a severe variant of FXTAS?', J Med Genet, 42(2), e14.
Jin, P., Zarnescu, D. C., Zhang, F., Pearson, C. E., Lucchesi, J. C., Moses, K. and Warren, S. T. (2003) 'RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila', Neuron, 39(5), 739-47.
Juncos, J. L., Lazarus, J. T., Graves-Allen, E., Shubeck, L., Rusin, M., Novak, G., Hamilton, D., Rohr, J. and Sherman, S. L. (2011) 'New clinical findings in the fragile X-associated tremor ataxia syndrome (FXTAS)', Neurogenetics, 12(2), 123-35.
Kaplan, E. S., Cao, Z., Hulsizer, S., Tassone, F., Berman, R. F., Hagerman, P. J. and Pessah, I. N. (2012) 'Early mitochondrial abnormalities in hippocampal neurons cultured from FMR1 pre-mutation mouse model', J Neurochem, 123(4), 613-21.
Kim, K., Taylor, S., Ta, B.T., Hagerman, P.J., & Berman, R.F. (2010) 'Expansion bias for transmission of expanded CGG-repeat murine FMR1 alleles', Proceedings of the 60th Annual Meeting of the American Society of Human Genetics, Washington, D.C.
Koldewyn, K., Hessl, D., Adams, J., Tassone, F., Hagerman, P. J., Hagerman, R. J. and Rivera, S. M. (2008) 'Reduced Hippocampal Activation During Recall is Associated with Elevated FMR1 mRNA and Psychiatric Symptoms in Men with the Fragile X Premutation', Brain Imaging Behav, 2(2), 105-116.
Kraan, C. M., Hocking, D. R., Bradshaw, J. L., Fielding, J., Cohen, J., Georgiou-Karistianis, N. and Cornish, K. M. (2013) 'Neurobehavioural evidence for the involvement of the FMR1 gene in female carriers of fragile X syndrome', Neurosci Biobehav Rev, 37(3), 522-47.
Leehey, M. A. (2009) 'Fragile X-associated tremor/ataxia syndrome: clinical phenotype, diagnosis, and treatment', J Investig Med, 57(8), 830-6.
Leehey, M. A., Berry-Kravis, E., Goetz, C. G., Zhang, L., Hall, D. A., Li, L., Rice, C. D., Lara, R., Cogswell, J., Reynolds, A., Gane, L., Jacquemont, S., Tassone, F., Grigsby, J., Hagerman, R. J. and Hagerman, P. J. (2008) 'FMR1 CGG repeat length predicts motor dysfunction in premutation carriers', Neurology, 70(16 Pt 2), 1397-402.
Leehey, M. A., Berry-Kravis, E., Min, S. J., Hall, D. A., Rice, C. D., Zhang, L., Grigsby, J., Greco, C. M., Reynolds, A., Lara, R., Cogswell, J., Jacquemont, S., Hessl, D. R., Tassone, F., Hagerman, R. and Hagerman, P. J. (2007) 'Progression of tremor and ataxia in male carriers of the FMR1 premutation', Mov Disord, 22(2), 203-6.
Linzenbold, W., Lindig, T. and Himmelbach, M. (2011) 'Functional neuroimaging of the oculomotor brainstem network in humans', Neuroimage, 57(3), 1116-23.
Manto, M. U. and Jissendi, P. (2012) 'Cerebellum: links between development, developmental disorders and motor learning', Front Neuroanat, 6, 1.
Metz, G. A. and Whishaw, I. Q. (2002) 'Cortical and subcortical lesions impair skilled walking in the ladder rung walking test: a new task to evaluate fore- and hindlimb stepping, placing, and co-ordination', J Neurosci Methods, 115(2), 169-79.
Moore, C. J., Daly, E. M., Schmitz, N., Tassone, F., Tysoe, C., Hagerman, R. J., Hagerman, P. J., Morris, R. G., Murphy, K. C. and Murphy, D. G. (2004) 'A neuropsychological investigation of male premutation carriers of fragile X syndrome', Neuropsychologia, 42(14), 1934-47.
Murray, A. (2000) 'Premature ovarian failure and the FMR1 gene', Semin Reprod Med, 18(1), 59-66.
Narcisa, V., Aguilar, D., Nguyen, D. V., Campos, L., Brodovsky, J., White, S., Adams, P., Tassone, F., Hagerman, P. J. and Hagerman, R. J. (2011) 'A Quantitative Assessment of Tremor and Ataxia in Female FMR1 Premutation Carriers Using CATSYS', Curr Gerontol Geriatr Res, 2011, 484713.
Peters, N., Kamm, C., Asmus, F., Holinski-Feder, E., Kraft, E., Dichgans, M., Bruning, R., Gasser, T. and Botzel, K. (2006) 'Intrafamilial variability in fragile X-associated tremor/ataxia syndrome', Mov Disord, 21(1), 98-102.
Primerano, B., Tassone, F., Hagerman, R. J., Hagerman, P., Amaldi, F. and Bagni, C. (2002) 'Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations', Rna, 8(12), 1482-8.
Qin, M., Entezam, A., Usdin, K., Huang, T., Liu, Z. H., Hoffman, G. E. and Smith, C. B. (2011) 'A mouse model of the fragile X premutation: effects on behavior, dendrite morphology, and regional rates of cerebral protein synthesis', Neurobiol Dis, 42(1), 85-98.
Sailer, U., Eggert, T., Ditterich, J. and Straube, A. (2000) 'Spatial and temporal aspects of eye-hand coordination across different tasks', Exp Brain Res, 134(2), 163-73.
Schluter, E. W., Hunsaker, M. R., Greco, C. M., Willemsen, R. and Berman, R. F. (2012) 'Distribution and frequency of intranuclear inclusions in female CGG KI mice modeling the fragile X premutation', Brain Res, 1472, 124-37.
Sellier, C., Freyermuth, F., Tabet, R., Tran, T., He, F., Ruffenach, F., Alunni, V., Moine, H., Thibault, C., Page, A., Tassone, F., Willemsen, R., Disney, M. D., Hagerman, P. J., Todd, P. K. and Charlet-Berguerand, N. (2013) 'Sequestration of DROSHA and DGCR8 by Expanded CGG RNA Repeats Alters MicroRNA Processing in Fragile X-Associated Tremor/Ataxia Syndrome', Cell Rep.
Sellier, C., Rau, F., Liu, Y., Tassone, F., Hukema, R. K., Gattoni, R., Schneider, A., Richard, S., Willemsen, R., Elliott, D. J., Hagerman, P. J. and Charlet-Berguerand, N. (2010) 'Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients', Embo J, 29(7), 1248-61.
Sherman, S. L. (2000) 'Premature ovarian failure in the fragile X syndrome', Am J Med Genet, 97(3), 189-94.
Simon, T. J. (2007) 'Cognitive characteristics of children with genetic syndromes', Child Adolesc Psychiatr Clin N Am, 16(3), 599-616.
Simon, T. J. (2008) 'A new account of the neurocognitive foundations of impairments in space, time and number processing in children with chromosome 22q11.2 deletion syndrome', Dev Disabil Res Rev, 14(1), 52-8.
Simon, T. J. (2011) 'Clues to the foundations of numerical cognitive impairments: evidence from genetic disorders', Dev Neuropsychol, 36(6), 788-805.
Spath, M. A., Feuth, T. B., Smits, A. P., Yntema, H. G., Braat, D. D., Thomas, C. M., van Kessel, A. G., Sherman, S. L. and Allen, E. G. (2011) 'Predictors and risk model development for menopausal age in fragile X premutation carriers', Genet Med, 13(7), 643-50.
Tassone, F., Beilina, A., Carosi, C., Albertosi, S., Bagni, C., Li, L., Glover, K., Bentley, D. and Hagerman, P. J. (2007) 'Elevated FMR1 mRNA in premutation carriers is due to increased transcription', Rna, 13(4), 555-62.
Tassone, F., Greco, C. M., Hunsaker, M. R., Seritan, A. L., Berman, R. F., Gane, L. W., Jacquemont, S., Basuta, K., Jin, L. W., Hagerman, P. J. and Hagerman, R. J. (2012) 'Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS', Genes Brain Behav.
Van Dam, D., Errijgers, V., Kooy, R. F., Willemsen, R., Mientjes, E., Oostra, B. A. and De Deyn, P. P. (2005) 'Cognitive decline, neuromotor and behavioural disturbances in a mouse model for fragile-X-associated tremor/ataxia syndrome (FXTAS)', Behav Brain Res, 162(2), 233-9.
van der Vaart, T., van Woerden, G. M., Elgersma, Y., de Zeeuw, C. I. and Schonewille, M. (2011) 'Motor deficits in neurofibromatosis type 1 mice: the role of the cerebellum', Genes Brain Behav, 10(4), 404-9.
Vianna-Morgante, A. M., Costa, S. S., Pares, A. S. and Verreschi, I. T. (1996) 'FRAXA premutation associated with premature ovarian failure', Am J Med Genet, 64(2), 373-5.
Wenzel, H. J., Hunsaker, M. R., Greco, C. M., Willemsen, R. and Berman, R. F. (2010) 'Ubiquitin-positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation', Brain Res, 1318, 155-66.
Whishaw, I. Q., Pellis, S. M., Gorny, B. P. and Pellis, V. C. (1991) 'The impairments in reaching and the movements of compensation in rats with motor cortex lesions: an endpoint, videorecording, and movement notation analysis', Behav Brain Res, 42(1), 77-91.
Willemsen, R., Hoogeveen-Westerveld, M., Reis, S., Holstege, J., Severijnen, L. A., Nieuwenhuizen, I. M., Schrier, M., van Unen, L., Tassone, F., Hoogeveen, A. T., Hagerman, P. J., Mientjes, E. J. and Oostra, B. A. (2003) 'The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome', Hum Mol Genet, 12(9), 949-59.
Willemsen, R., Levenga, J. and Oostra, B. (2011) 'CGG repeat in the FMR1 gene: size matters', Clin Genet, 80(3), 214-25.
Wong, L. M., Goodrich-Hunsaker, N. J., McLennan, Y., Tassone, F., Harvey, D., Rivera, S. M. and Simon, T. J. (2012) 'Young adult male carriers of the fragile X premutation exhibit genetically modulated impairments in visuospatial tasks controlled for psychomotor speed', J Neurodev Disord, 4(1), 26.
Xu, T., Yu, X., Perlik, A. J., Tobin, W. F., Zweig, J. A., Tennant, K., Jones, T. and Zuo, Y. (2009) 'Rapid formation and selective stabilization of synapses for enduring motor memories', Nature, 462(7275), 915-9.
Zeiler, S. R., Gibson, E. M., Hoesch, R. E., Li, M. Y., Worley, P. F., O'Brien, R. J. and Krakauer, J. W. (2013) 'Medial premotor cortex shows a reduction in inhibitory markers and mediates recovery in a mouse model of focal stroke', Stroke, 44(2), 483-9.